Discinesia ciliar primaria. Causa de infecciones respiratorias recurrentes: serie de tres casos
Leal-Jiménez, Elizabeth1; Avilés-Ramírez, Brenda Aidé1; Reyes-Rosales, Mariana1

2023, Número 1
2023; 82 (1)


Leal-Jiménez, Elizabeth1; Avilés-Ramírez, Brenda Aidé1; Reyes-Rosales, Mariana1
RESUMEN
La discinesia ciliar primaria es una enfermedad genética poco frecuente, autosómica recesiva, que afecta el movimiento ciliar. Las manifestaciones clínicas de la enfermedad pueden presentarse desde el nacimiento con un síndrome de dificultad respiratoria o durante la infancia con tos crónica productiva y rinosinusitis no estacional, infecciones respiratorias recurrentes que incluso requieran manejo intrahospitalario, por lo que se deberá considerar como parte del abordaje de estudio de infecciones recurrentes en el paciente pediátrico.PALABRAS CLAVE
discinesia ciliar, infecciones respiratorias, tos crónica.Introducción
La discinesia ciliar primaria (DCP) es una enfermedad genética poco frecuente, generalmente se presenta con un patrón de herencia autosómico recesivo que afecta el movimiento de los cilios, con afección principalmente de los genes DNAH5 y DNAl1.1 Se considera una enfermedad de inicio temprano sin predilección de género, etnia y/o raza, tiene una frecuencia de 1 en 10,000-20,000 recién nacidos vivos, con prevalencia incrementada hasta de 5% en pacientes con infecciones de vías respiratorias de repetición en países europeos.2
Los cilios son organelos que se encuentran en la superficie celular. Existen dos tipos de cilios: los inmóviles y los móviles; cada cilio está compuesto por un citoesqueleto, llamado axonema, compuesto de nueve dobletes de microtúbulos longitudinales que rodean un par central, conformando el 9 + 2. Cada doblete de microtúbulos periféricos tiene un brazo externo y un brazo interno de dineína, la cual contiene la proteína motriz del cilio.3 El 70% de los pacientes con DCP tienen una variante patogénica en algún gen relacionado con el funcionamiento de los cilios, provocando cambios estructurales y funcionales. En el caso de la DCP, la afección se encuentra en cilios móviles encargados de la acción de barrido, movilizando fluidos y residuos de forma unilateral. Al encontrarse un defecto en este movimiento, el paciente presentará un aclaramiento mucociliar anómalo, lo cual se manifiesta clínicamente en el paciente, presentando síntomas como infecciones de vías respiratorias de repetición en niños, casos de infertilidad en hombres y mujeres al afectar el movimiento de los espermatozoides y movimiento ciliar de las trompas de uterinas, respectivamente.4-6 Asimismo, se han reportado casos con presentación clínica desde el período neonatal con atelectasias e infecciones respiratorias recurrentes, acompañadas en 50% por situs inversus.
Forman parte del diagnóstico diferencial patologías como fibrosis quística, inmunodeficiencias, aspiración pulmonar, asma e infecciones de vías respiratorias recurrentes (sinusitis, otitis, rinofaringitis, neumonías, entre otras); sin embargo, se han identificado cuatro características clave para el diagnóstico de DCP: 1) tos productiva (húmeda) durante todo el año; 2) rinosinusitis no estacional diaria y de inicio a temprana edad; 3) 80% antecedente de síndrome de dificultad respiratoria al nacimiento; y 4) defectos de lateralidad.7
A continuación, se exponen tres casos de pacientes con diagnóstico de DCP mediante microscopia electrónica.
Presentación de los casos
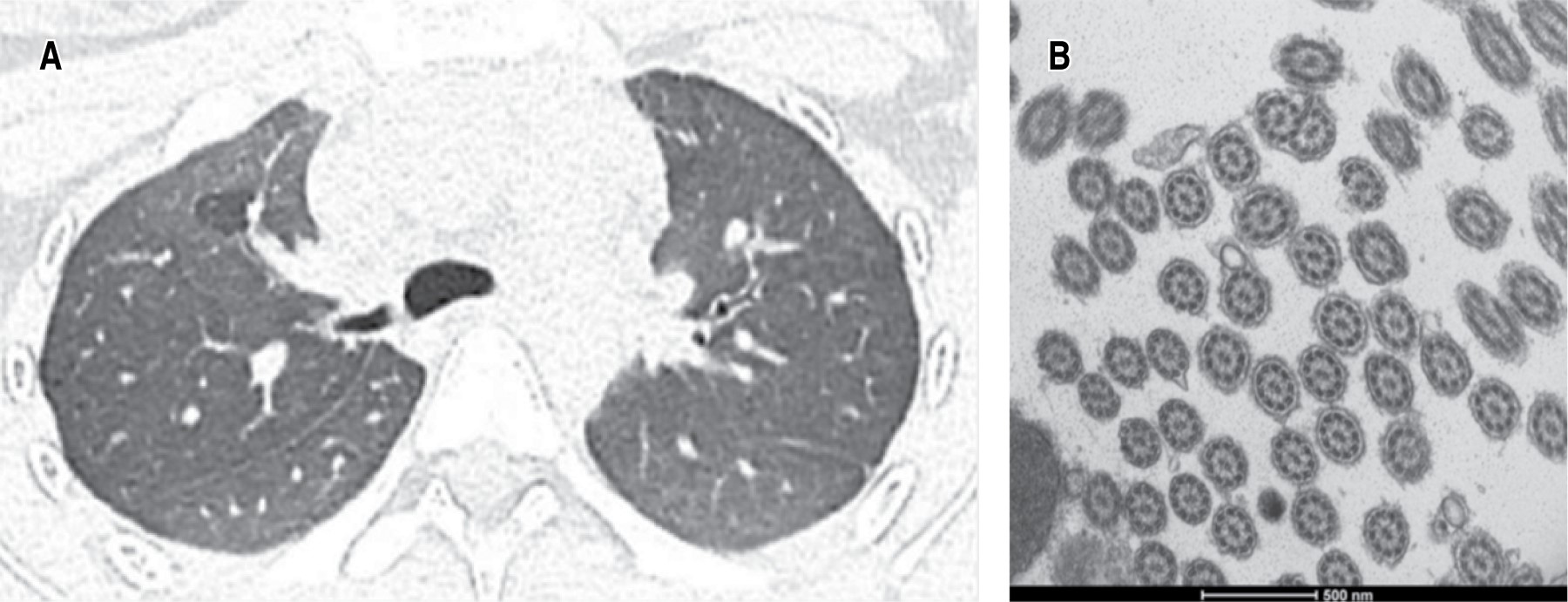
Caso 1: femenino de ocho años con antecedente de infecciones de vías respiratorias recurrentes desde el nacimiento, tres eventos de neumonía, dos de influenza y tres cuadros de otitis. Se realizó diagnóstico de asma por espirometría con obstrucción leve con respuesta positiva a broncodilatador, persistiendo con tos húmeda productiva a pesar de dosis altas de tratamiento de control de asma. De forma recurrente, presenta sibilancias y estertores gruesos. Se realizó abordaje con tomografía de tórax de alta resolución (TACAR) con presencia de bronquiectasias cilíndricas (Figura 1A), fueron descartadas patologías como fibrosis quística, inmunodeficiencias, reflujo gastroesofágico y aspergilosis broncopulmonar. Se realizó broncoscopia flexible para toma de biopsia de cilio (endobronquial), la cual se reportó con epitelio endobronquial con alteraciones ultraestructurales y de membrana compatible con discinesia ciliar (procesada por microscopia electrónica), con ausencia de brazos de dineína que corresponde a DCP tipo I según la clasificación de Barlocco y colaboradores8 (Figura 1B). Por parte del servicio de genética se realizó estudio molecular de panel de 538 genes por secuenciación de nueva generación para ciliopatías, el cual informó que la paciente es heterocigota compuesta para dos variantes en el gen DNAH5, la variante c.2578-2A>T está reportada previamente como patogénica; la variante c.1981C>A (p.Arg661Ser) está clasificada como de significado incierto (Figura 1C). Se establece diagnóstico de DCP y, con ello, la reducción de dosis de inhalador hasta el retiro; se inicia terapia de rehabilitación pulmonar y manejo de forma multidisciplinaria con los servicios de otorrinolaringología, audiología y cardiología pediátrica. Se descartó situs inversus.
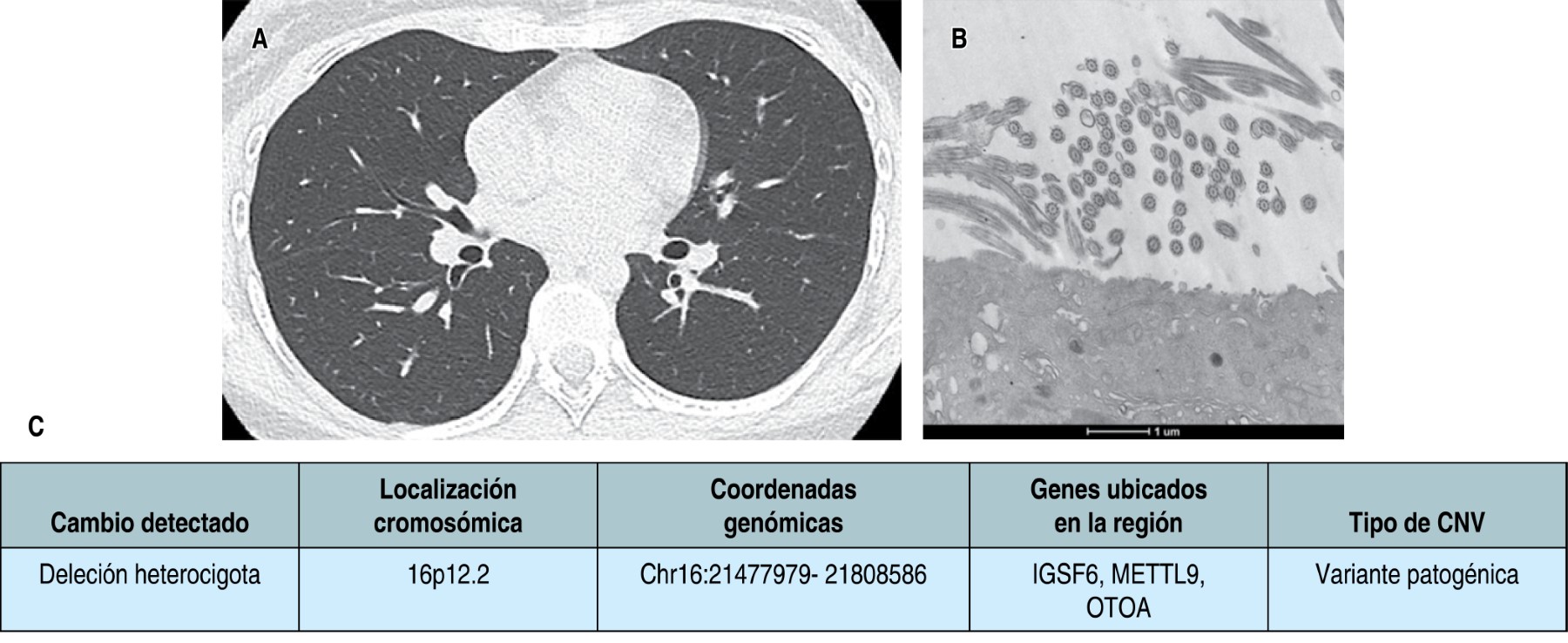
Caso 2: femenino de 16 años, con infecciones respiratorias de repetición desde los tres años, tratada previamente por asma de difícil control, todas sus espirometrías con obstrucción leve sin respuesta a broncodilatador; se realizó abordaje por la persistencia de la sintomatología respiratoria (tos productiva, abundantes secreciones en vía aérea y cuadros de sibilancias recurrentes). TACAR con presencia de bronquiectasias cilíndricas (Figura 2A). Se realizó broncoscopia flexible con toma de biopsia endobronquial para microscopia electrónica, en la cual se reportan cambios compatibles con DCP tipo V de la clasificación de Barlocco y colaboradores,8 correspondientes a fusión ciliar (Figura 2B). Se solicita panel genético de ciliopatías, el cual señala deleción heterocigota del cromosoma 16p12.2 variante patogénica (Figura 2C). Se inició terapia de rehabilitación pulmonar, se disminuyó dosis de inhaladores hasta el retiro, con mejoría de los síntomas respiratorios. Actualmente sólo se encuentra con fisioterapia pulmonar. Se inició manejo multidisciplinario a través de los servicios de genética, cardiología y otorrinolaringología. Se descartó situs inversus.
Caso 3: femenino de 14 años con diagnóstico de tos crónica y síndrome supurativo. Al nacimiento presentó infección de vía aérea. Durante su infancia con infecciones recurrentes de faringoamigdalitis, otitis media de repetición, tratamiento quirúrgico de amigdalectomía y colocación de tubos de ventilación; falla de medro. Presenta eventos recurrentes de broncoespasmo, tos productiva, sin requerimiento de hospitalización. Se solicitó TACAR, con presencia de bronquiectasias y engrosamiento peribronquial (Figura 3A), espirometría con broncodilatador normal sin reversibilidad, serie esofagogastroduodenal con reflujo grado I. Se realizó broncoscopia con toma de biopsia endobronquial para microscopia electrónica; reporta alteraciones compatibles con DCP, con alteración en los microtúbulos, ausencia de dobletes periféricos (Figura 3B), DCP tipo III de la clasificación de Barlocco y colaboradores.8 Se solicita panel genético.
Discusión
La mayoría de los pacientes con DCP presentan manifestaciones clínicas a temprana edad; sin embargo, el diagnóstico se retrasa hasta una edad media de cuatro a seis años secundario al bajo índice de sospecha.9 Con la finalidad de realizar un diagnóstico en edades más tempranas, se debe sospechar esta patología en pacientes con síntomas respiratorios recurrentes o crónicos como tos húmeda, rinorrea, sinusitis, otitis media y/o presencia de bronquiectasias, con el antecedente de recién nacido a término con ingreso a Unidad de Cuidados Intensivos Neonatales por patología pulmonar, cardiopatía congénita o alteraciones de la lateralidad.10 En nuestra serie de casos, observamos que las tres pacientes presentaron cuadros recurrentes de infecciones de vía respiratoria, tratados inicialmente como asma de difícil control. Asimismo, el 100% presentó alteraciones radiológicas como engrosamiento peribronquial y bronquiectasias, se utilizó TACAR en todos los casos.
Para el diagnóstico de DCP no existe una única prueba diagnóstica, por lo que se han propuesto escalas predictivas como PICADAR utilizadas en combinación con distintos métodos de estudio. En nuestra institución contamos con broncoscopia flexible, por lo cual se decidió realizar el diagnóstico mediante este método; se subrogó la interpretación por microscopia electrónica en el Instituto Nacional de Ciencias Médicas y Nutrición "Salvador Subirán", ya que son pocos los centros que cuentan con este recurso. Nuestra institución cuenta con servicio de genética, por lo que se solicitó estudio molecular para ciliopatías. En la actualidad, existen en centros especializados otros métodos como la fracción exhalada de óxido nítrico; en nuestro centro, sin embargo, no contamos con este recurso, lo cual resulta una limitante para este estudio.
Dentro de los estudios complementarios para medir la función pulmonar se solicitó espirometría con broncodilatador, la cual puede ser normal durante la primera infancia, llegando, conforme a la evolución del cuadro, hasta obtener cierto grado de obstrucción del flujo de aire.
Al presente, no existe un tratamiento para corregir la disfunción ciliar. El tratamiento consistente es la fisioterapia pulmonar para mejorar el aclaramiento mucociliar y evitar sobreinfección bacteriana, por lo que se recomienda el inicio temprano de antibioticoterapia ante una infección de vías respiratorias.11 Es importante el manejo multidisciplinario.
Conclusiones
Los pacientes con DCP presentan un cuadro clínico variable, con manifestaciones de leves a moderadas, por lo regular de inicio temprano y diagnóstico tardío, afectando la calidad de vida del paciente. Al ser una patología de herencia autosómica recesiva, se requiere asesoramiento genético a los padres, ya que no existe un tratamiento definitivo para recuperar el movimiento del cilio afectado. En nuestro estudio se observó que aún tenemos limitantes para lograr un diagnóstico temprano debido a la heterogeneidad de los síntomas y la falta de recursos, como por ejemplo la microscopia electrónica y la fracción exhalada de óxido nítrico, pero es sumamente importante aplicar escalas de sospecha diagnóstica e iniciar de forma temprana el abordaje para descartar otras patologías. Se planea aplicar el estudio y dar seguimiento de forma prospectiva a estos pacientes para tener un mayor conocimiento sobre pronóstico y calidad de vida.
AFILIACIONES
1Centro Médico Naval SEMAR. Ciudad de México, México.Conflicto de intereses: los autores declaran no tener conflicto de intereses.
REFERENCIAS
Shapiro AJ, Davis SD, Polineni D, Manion M, Rosenfeld M, Dell SD, et al.; American Thoracic Society Assembly on Pediatrics. Diagnosis of primary ciliary dyskinesia. An official American Thoracic Society Clinical Practice Guideline. Am J Respir Crit Care Med. 2018;197(12):e24-e39. Available in: https://doi.org/10.1164/rccm.201805-0819st