La fibrosis pulmonar vista desde sus primeros casos publicados en México
Buendía-Roldán, Ivette1

2024, Suplemento 1
2024; 83 (S1)


Buendía-Roldán, Ivette1
RESUMEN
PALABRAS CLAVE
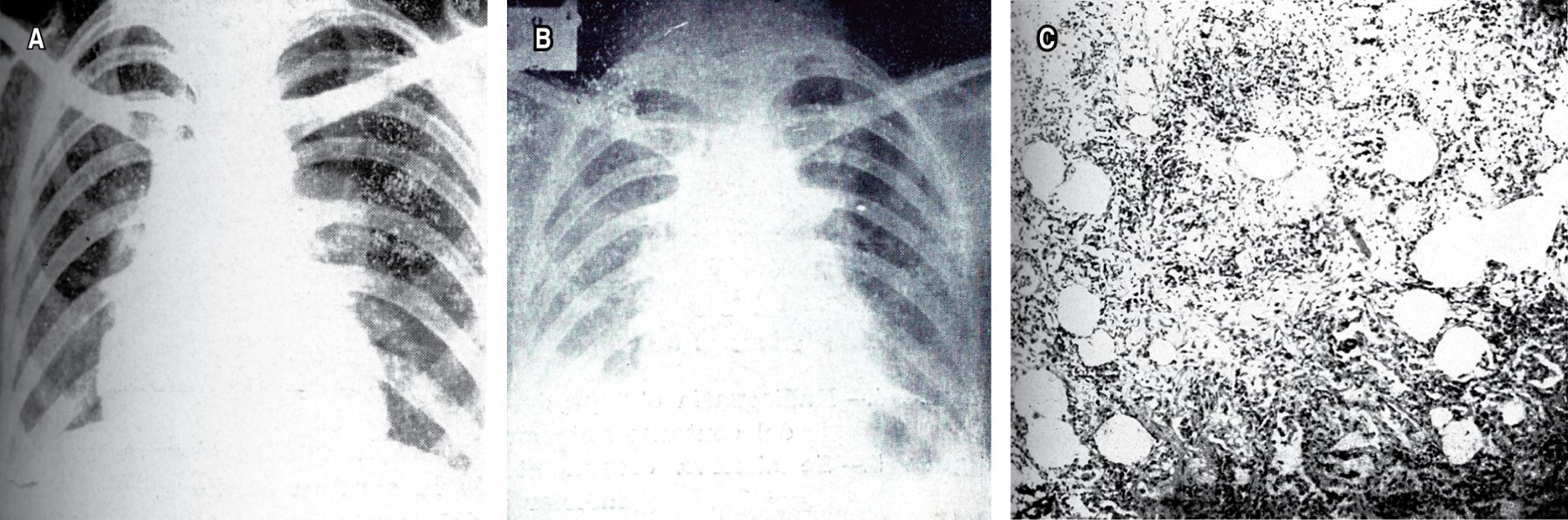
.Las enfermedades pulmonares intersticiales difusas (EPID) han sido siempre un enigma para el personal médico; se debe en parte a la variedad de etiologías relacionadas con ellas, así como a los cambios histopatológicos que se reportan en las biopsias o autopsias y los escasos datos, clínicos o de las pruebas funcionales respiratorias, que comparten todas ellas. Las primeras publicaciones sobre fibrosis pulmonar difusa en NCT (Revista Mexicana de Tuberculosis) corresponden a dos casos descritos con detalle en el aspecto clínico y en sus características anatomopatológicas de autopsia. El primero de ellos data de 1956 y se titula: "Fibrosis pulmonar intersticial difusa" del Dr. F. Marroquín de la Unidad de Patología de la Escuela de Medicina de la UNAM, en el Hospital General de México.1 El segundo caso fue publicado en 1961: "Fibrosis intersticial difusa. Caso clínico-patológico", por los doctores José Vargas de la Cruz, Enrique Milán Reyes y Arturo Tinoco Hernández del Servicio de Anatomía Patológica del Hospital de La Raza del IMSS.2 Desde entonces y en ambas publicaciones, los autores destacan la poca correlación entre los datos clínicos y los cambios macro y microscópicos (Figuras 1 y 2), más aún, la variabilidad en edades, lo avanzado de los casos al momento del diagnóstico y la falta de asociación con las causas conocidas de la enfermedad, por ejemplo, las exposiciones ambientales o las infecciones, así como la nula respuesta a los tratamientos implementados; todo esto incrementaba la incertidumbre ante estas enfermedades.
En un intento por tener definiciones claras, se han realizado diferentes clasificaciones de enfermedades pulmonares intersticiales. En 2013, la última actualización de la clasificación internacional de las neumonías intersticiales idiopáticas,3 las divide en neumonías intersticiales idiopáticas mayores, neumonías intersticiales idiopáticas raras y las neumonías intersticiales idiopáticas no clasificables. A partir de entonces, se han realizado dos actualizaciones, en específico para la fibrosis pulmonar idiopática (FPI), debido a su evolución siempre progresiva y a su alta letalidad. En estas actualizaciones se ha propuesto que se deben reunir una serie de datos clínicos, tomográficos y morfológicos que permitan realizar un diagnóstico adecuado y en poco tiempo. Resalta la importancia del diagnóstico multidisciplinario, ya que es una enfermedad que requiere se descarten otras causas que expliquen la presencia de fibrosis pulmonar, como sería la neumonitis por hipersensibilidad o las enfermedades del tejido conectivo.4 En la última actualización publicada en 2022,5 se agrega por primera vez al léxico de las EPID, el término fibrosis pulmonar progresiva (FPP); que define un punto a partir del cual ya se han activado diferentes vías fisiopatogénicas fibróticas y, por lo tanto, independientemente del origen de la enfermedad intersticial, el desenlace va a ser similar al de la FPI. Para denominarse FPP se deben cumplir dos de tres criterios: incremento de los síntomas respiratorios, diminución ≥ 5% en la capacidad vital forzada (FVC, por sus siglas en inglés) y/o ≥ 10% en difusión pulmonar de monóxido de carbono (DLCO), durante un año de seguimiento y con la aparición o progresión de las lesiones tomográficas preexistentes. Cabe aclarar que este término no se utiliza para la FPI, ya que esta última siempre es progresiva.
Con respecto a la exploración física, el artículo del Dr. Marroquín deja claro, desde 1956, la importancia de la auscultación del tórax para la identificación de estertores pulmonares que se presentaban en 72% de los 43 casos de EPID reportados hasta entonces. Este signo sigue siendo de utilidad para la sospecha diagnóstica temprana de los pacientes,6 incluso se ha relacionado con predicciones de patrones radiológicos específicos en EPID fibrótica.7
Es relevante desde los primeros reportes y se menciona con relativa seguridad, que el fenómeno fundamental es la producción de tejido conectivo y las alteraciones vasculares-cardíacas. Un parteaguas en el entendimiento de las EPID, sin duda fue la publicación de la entonces hipótesis de la patogénesis de la FPI,8 que la diferenció del resto de las enfermedades intersticiales. Durante más de 20 años, un número creciente de estudios y publicaciones, entre ellas del grupo del Dr. Moisés Selman en el INER, han corroborado el papel de las células epiteliales, los fibroblastos, la matriz extracelular, así como la importancia del envejecimiento en la fisiopatogenia y la evolución de los pacientes. La importancia de estos estudios ha sido a tal grado que, desde hace casi 10 años, se aprobaron dos medicamentos llamados "antifibrosantes": la pirfenidona y el nintedanib, con la indicación inicial de FPI; por primera vez, ambos medicamentos demostraron que eran capaces de enlentecer la caída de la FVC en estos pacientes.9,10 Actualmente, se están utilizando en diferentes ensayos clínicos para comprobar su eficacia en otras EPID fibróticas e incluso en la FPP. En algunos países se cuenta con trasplante pulmonar que es una excelente alternativa terapéutica para pacientes con formas avanzadas de las EPID; sin embargo, no es accesible en muchos otros países. El avance tecnológico ha permitido que ya se realice a través de cirugía robótica, con excelentes resultados y un cambio de vida, tanto para los pacientes como sus familiares.
A lo largo de estos años, los estudios para entender la fisiopatología y el diagnóstico de la fibrosis pulmonar han avanzado en relación con los progresos tecnológicos; no sólo con reportes de casos clínicos, también con estudios con múltiples enfoques, como: la recapitulación aberrante de programas de desarrollo,11 el papel de las metaloproteínas de matriz que favorecen un fenotipo profibrótico,12 la secuenciación unicelular de sangre periférica,13 así como la incesante búsqueda de biomarcadores moleculares en muestras sanguíneas y en aliento14 y, más recientemente, el uso de la tecnología basada en inteligencia artificial para el análisis de la tomografía de tórax.15 A pesar de toda esta generación de conocimiento, las enfermedades intersticiales siguen siendo un enigma, principalmente aquellas llamadas huérfanas y las de aparición en la niñez. Sin lugar a dudas, este tema seguirá siendo de interés, no sólo para la investigación clínica y básica, sino también para la investigación traslacional, principalmente ante la necesidad de encontrar tratamientos específicos que consigan detener el proceso fibrótico e, idealmente, logren regenerar los sitios lesionados.
Hasta el momento, un objetivo sin cumplir sigue siendo lograr una sospecha diagnóstica de la EPID desde la primera atención médica del paciente y alcanzar su diagnóstico en las fases incipientes de la enfermedad, particularmente con apoyo y tratamiento multidisciplinario; todo ello con la intención de poder ejercer una medicina personalizada en cada paciente y contar con tratamientos específicos, tanto farmacológicos como no farmacológicos, que permitan controlar y mejorar su calidad de vida.
AFILIACIONES
1Instituto Nacional de Enfermedades Respiratorias Ismael Cosío Villegas. Ciudad de México, México.Conflicto de intereses: el autor declara no tener conflicto de intereses.
REFERENCIAS
Travis WD, Costabel U, Hansell DM, King TE Jr, Lynch DA, Nicholson AG, et al.; ATS/ERS Committee on Idiopathic Interstitial Pneumonias. An official American Thoracic Society/European Respiratory Society statement: update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2013;188(6):733-748. Available in: https://doi.org/10.1164/rccm.201308-1483st
Raghu G, Remy-Jardin M, Myers JL, Richeldi L, Ryerson CJ, Lederer DJ, et al.; American Thoracic Society, European Respiratory Society, Japanese Respiratory Society, and Latin American Thoracic Society. Diagnosis of idiopathic pulmonary fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am J Respir Crit Care Med. 2018;198(5):e44-e68. Available in: https://doi.org/10.1164/rccm.201807-1255st
Raghu G, Remy-Jardin M, Richeldi L, Thomson CC, Inoue Y, Johkoh T, et al. Idiopathic pulmonary fibrosis (an update) and progressive pulmonary fibrosis in adults: an official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 2022;205(9):e18-e47. Available in: https://doi.org/10.1164/rccm.202202-0399st
Selman M, King TE, Pardo A; American Thoracic Society; European Respiratory Society; American College of Chest Physicians. Idiopathic pulmonary fibrosis: prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann Intern Med. 2001;134(2):136-151. Available in: https://doi.org/10.7326/0003-4819-134-2-200101160-00015