Eicosanoids as regulators of inflammation and immunology process during pulmonary tuberculosis
Escalona-Sarabia, Ana Luisa1; Juárez, Esmeralda2

2022, Number 2
2022; 81 (2)


Escalona-Sarabia, Ana Luisa1; Juárez, Esmeralda2
ABSTRACT
Human tuberculosis (TB) is a public health problem. Although it is a curable disease, the actual treatment protocol is long and difficult to maintain. Because of the limit progress of new therapies during the latest years, host direct therapies have taken more interest, being one of the proposals the intervention on the eicosanoids' metabolic route. Eicosanoids are local chemical mediators that promote or limit inflammation progress. During TB, hyperinflammation generates damage to the pulmonary parenchyma with the subsequent deterioration of respiratory function. Despite the importance of this circuit, reports about the utility in tuberculosis sometimes are controversial or not conclusive. With the aim of knowing and integrate the information published, in this review we search and analyze different studies that look forward to defining the eicosanoids' role on M. tuberculosis infection. For this, we review the role of eicosanoids post-infection in vivo or in vitro, and the modification of their metabolic route before or after infection. We also propose an algorithm to optimize the future investigations of eicosanoids and their utility as therapeutic target during TB.KEYWORDS
tuberculosis, eicosanoids, host-directed therapies.Introduction
Human tuberculosis (TB), caused by Mycobacterium tuberculosis, is one of the 10 leading death causes in the world.1 In Mexico, more the 20,000 new cases are reported each year, mostly in the pulmonary form.2,3 Chronic inflammation in the lungs can cause damage to the parenchyma and deterioration of the respiratory function,4,5 making the inflammatory circuit a therapeutic target to preserve pulmonary function. Consequently, host-directed therapies such as cytokines modulators (IFN-γ and TNF-α), anti-fibrotic, or molecules that activate macrophages are ideal because of their ability to modulate the immune system and limit post-infection tissue damage.6,7
Intervening the metabolic pathway of eicosanoids is a therapeutic option to prevent inflammation and preserve pulmonary function. Eicosanoids are lipids derived from polyunsaturated fatty acids (PUFA) that are obtained from the intake of omega-6 or ω-6 fatty acids (arachidonic acid [AA]) and omega-3 or ω-3 (eicosapentaenoic acid [EPA] and docosahexaenoic acid [DHA]). These lipids are synthesized enzymatically by lipoxygenases (LOX) and/or cyclooxygenases (COX) in leukocytes, endothelial cells and platelets.7 There are two main families of PUFA with antagonistic activities, pro-inflammatory and pro-resolution.
The pro-inflammatory eicosanoids include thromboxanes, prostaglandins and prostacyclins synthesized by COX-2, and leukotrienes, synthesized by 5 and 15-LOX from AA. Pro-inflammatory eicosanoids are chemoattractant of neutrophils, activate phagocytosis in alveolar macrophages,8 mediate the cells trafficking,9 and induce necrosis, edema, increase blood flow and production of pro-inflammatory cytokines. Pro-resolution eicosanoids, or specialized pro-resolution lipid mediators (SPMs), are synthesized by 5 and 15-LOX and include lipoxins, resolvins, protectins and maresins.10 These lipids limit the flow of neutrophils, block the production of reactive oxygen species, induce apoptosis and increase the macrophage phagocytic activity favoring the return of homeostasis and cell regeneration.11,12
As important as this circuit is for the respiratory infection, its contribution in the inflammatory regulation in TB needs to be reviewed in terms of published experimental models. In this revision, we will consider the molecular mechanisms of eicosanoids involved in the regulation of inflammation during TB in animal models; we will observe that some phenomena observed in animal models cannot be replicated in humans and vice versa; and we will analyze the validity of using host-directed therapies based on the molecular mechanisms that eicosanoids regulate.
Experimental models used to study the role of eicosanoids in tuberculosis
To define the role that eicosanoids play in M. tuberculosis infection we established three experimental designs: 1) post-infection eicosanoid quantification; 2) therapeutic intervention in vivo with polyunsaturated fatty acids (PUFA) before infection; and 3) therapeutic intervention with PUFA post-infection.
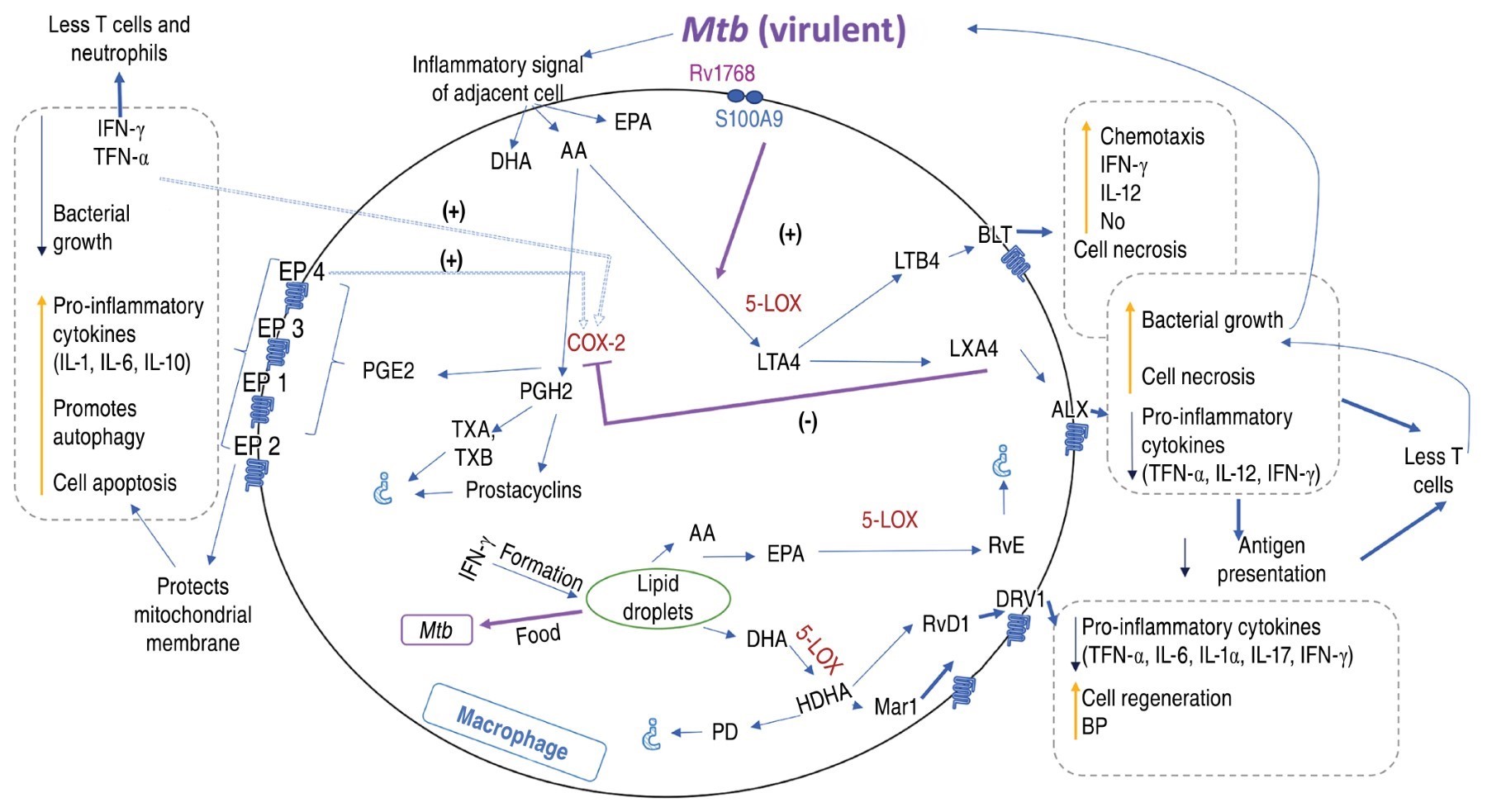
Post-infection eicosanoid quantification. Animal models infected in vivo with virulent strains of M. tuberculosis (H37Rv, Erdman or HN878), as well as samples of patients, reveal changes in eicosanoids levels during and after TB infection that correlate with the severity of the disease.13,14 The most studied eicosanoids are derived from AA: PGE2, lipoxin A4 (LXA4) and LTB4. The presence of LXA4 and LTB4 is associated with necrosis and tissue damage,12 while the presence of PGE2, or its receptor EP2, have opposite effects and are associated with infection resistance.12-14 Increase of LXA4 is associated with a greater susceptibility to disease and PGE2 with a protective response. In the pulmonary tissue of patients with pulmonary TB, the presence of AA and leukotriene A4 hydrolase (LTA4H) are observed in necrotic centers and presence of cyclooxygenases at the periphery of the lesions are observed.15 Patients with pulmonary TB, multidrug-resistent (MDR) TB or latent TB have higher circulating amounts of PGE2, LTB4 and LXA4 than a healthy person.16-18
Although other eicosanoids were measured in some studies, such as maresin (Mar1, Mar2), resolvins RvD (RvD1-6) and RvE (RvE1-4); prostaglandins (PGF2, PGD2), protectins (PD1) and eicosanoid precursors 12-HETE or 15-HETE,19-21 none of these are relevant for TB individually.Rather, the effects of eicosanoids depend on their relative contribution.22 For example, an increase in serum LTB4/Mar1 ratio distinguishes patients from healthy individuals,19 the LTB4/LXA4 ratio decreases post-treatment,13 the connections between SPM and pro-inflammatory lipids are higher in TB-DM patients compared to TB-only patients,20 and PGE2 levels depend on the LTB4/LXA4 ratio.23
The relationship between PGE2 and LXA4 is antagonistic in macrophage cultures, LXA4 induces necrosis and PGE2 induces apoptosis for cell protection against infection.24-26 As in in vivo determinations, increased LXA4 is associated with increased susceptibility to infection, increased inflammation and bacillary burden. In addition, M. tuberculosis infection increases the release of AA in macrophages and its transformation into LXA4 mediated by 5-LOX.24 Inhibition of LXA4 synthesis protects against necrosis,12 and therefore its induction seems to be a survival strategy of the mycobacterium. Regarding SPM, RvD1 and Mar1, they induce anti-inflammatory mechanisms and restore the synthesis of antimicrobial peptides in human macrophages infected with the virulent strain M. tuberculosis H37Rv.25 The lack of measurement of other eicosanoids prevents us from knowing the real impact of the inflammation-resolution circuit during M. tuberculosis infection.
Therapeutic interventions performed in vivo prior to infection. Research in which eicosanoids are administered or therapeutics that modify their metabolic pathways are applied prior to infection can explain the mechanisms involved during the disease process. The most common interventions are the use of diets enriched with omega-3 and omega-6 fatty acids, diets deficient in these fatty acids, and drugs that inhibit eicosanoid synthesis.
Mice treated with ω-6 supplemented diets26,27 and guinea pigs fed with ω-3 supplements28 show increased bacterial load, but BALB/c and C3HeB/FeJ mice fed omega-3 enriched diets show reduced bacterial load and reduced amounts of pro-inflammatory cytokines released into the local environment.26,29,30 In humans, a longitudinal-prospective study revealed that there was a higher risk of developing TB at higher cholesterol intake and a lower risk of developing TB at higher intakes of ω-3 and ω-6 of marine origin.31 However, due to their antagonistic biological effects, it is difficult to reach a conclusion when consumption is varied and occurs prior to infection.
Drug intervention includes 5-LOX or COX-2 inhibitors to manipulate the metabolic pathway. Inhibiting 5-LOX has negative effects because it increases susceptibility to infection by decreasing the number of leukocytes and increasing bacterial load.32-34 The molecular mechanism involved is uncertain, since 5-LOX participates in the synthesis of LTB4 and LXA4 from AA, both with antagonistic effects, and also in the production of resolvins from DHA and EPA, which are pro-resolution. In cases where 5-LOX inhibitor was administered, the absence of resolvins, and not LTB4, could be the reason for the lack of infection control.
COX-2 inhibition has benefits such as a decrease in bacterial load, the size and presence of granulomas and mortality.19,32-35 COX-2 inhibition involves blocking prostaglandins, prostacyclins and thromboxanes, whose biological effects on TB have not been explored. This strategy has prophylactic potential for people exposed by close contact with patients. However, in most of these trials, treatments were administered before infection and continued during infection, so it is difficult to know whether the result is due to activation of pre-infection mechanisms or to their post-infection maintenance.
Therapeutic interventions performed post-infection. Post-infection interventions include direct supplementation of eicosanoids (Table 1) or pharmacological inhibition of their synthesis (Table 2). Schemes using dietary changes or agonists of PGE2, LTB4 or other eicosanoids post-infection are scarce. In general, direct supplementation with eicosanoids reduces the production of pro-inflammatory cytokines, influences pathogenicity19 or confers protection.36,37 Paradoxically, PGE2 is a pro-inflammatory with immunosuppressive effects.12 In addition, RvD1 and Mar1 have anti-inflammatory effects without detriment to antimicrobial mechanisms.25
On the other hand, eicosanoids of both types are produced during infection,38-40 and diets supplemented with DHA/EPA have anti-inflammatory effects,41,42 but the relationship between the two phenomena is unknown. Further research is needed to know the true potential of DHA/EPA supplementation during TB in humans.
Pharmacological inhibition of eicosanoid synthesis during M. tuberculosis infection (Table 2) relies mainly on the in vivo, in vitro and ex vivo use of COX-2 inhibitors (niflumic acid [NA], aspirin, celecoxib, etoricoxib or ibuprofen). This inhibition generally causes reduced inflammation by decreasing cytokine production, reduced tissue damage, and increased survival. Controversially, inhibiting COX-2 may increase the bacterial load when treatment is applied in early stages of infection,43 probably due to the immunosuppressive effects of PGE2,12 whereas the late use of these inhibitors allows a better resolution of the disease, with reduction of bacterial load and inflammation, which protects against tissue damage.33,44-47 COX-2 inhibition needs to be taken with caution, as its immunosuppressive effects may affect the patient during the early stages of the disease and further studies are required to determine its efficacy in TB.
Molecular mechanisms associated to metabolism of eicosanoids in M. tuberculosis infection
During the first hours of infection, AA of nuclear and plasma membrane is processed by COX-1 and COX-2 into PGH2, which it is converted to PGE2 by cPGES, mPGES-1 or PGES-2.48 In infected macrophages, PGE2 production correlates with decreased phagocytosis, nitric oxide production, prevention of necrosis, increased pro-inflammatory cytokines and induction of apoptosis, protecting the mitochondrial membrane, promoting plasma membrane repair and enhancing the control of innate immunity to mycobacterial infection.14,18,49 PGE2 also activates autophagy by enhancing bacterial elimination in autophagolysosomes. Autophagy, in turn, controls inflammation by regulating innate immune signaling, modulating the secretion of immune mediators and eliminating endogenous agonists from the inflammasome.50
After the first 24 hours post-infection, there is an increased production of LXA437 in macrophages, which causes a change in AA metabolism mediated by 5-LOX.24 Increased LXA4 levels are associated with reduced necrosis, bacillary burden, pro-inflammatory cytokines, vascular permeability, polymorphonuclear leukocyte (PMN) entry to the sites of inflammation and Th1-type protective response.14,15,18
The effect of 5-LOX on AA also causes an increase in LTB4. During the early stages of inflammation, only mesothelial cells and macrophages are able to release LTB4 into the pleural space in response to an initial inflammatory stimulus. Once the inflammatory process is established, other cells, such as neutrophils, produce LTB4 amplifying the inflammatory process.51 LTB4, in turn, induces necrosis, increased nitric oxide production and chemotaxis (Figure 1).
On the other hand, AA is also metabolized in thromboxanes and prostacyclins,19,52 but these have not been associated with TB. Other membrane PUFA that are metabolized during inflammation and cellular stress are DHA and EPA. The conversion of omega-6 and omega-3 precursors into PUFA is controlled by fatty acid desaturase enzymes (FADS) 1 and 2.36 Subsequently, these are transformed by 5-LOX into maresin, protectins and resolvins. Although they have been reported during TB,35,53,54 the involvement of these eicosanoids in the disease process is unknown, with the exception of resolvin D1 (RvD1) and maresin 1 (Mar1) which contribute to the control of M. tuberculosis infection in vitro by increasing bactericidal permeability-increasing protein (BPI) and cell regeneration.25
Scope of interventional therapies of the eicosanoid metabolic pathway
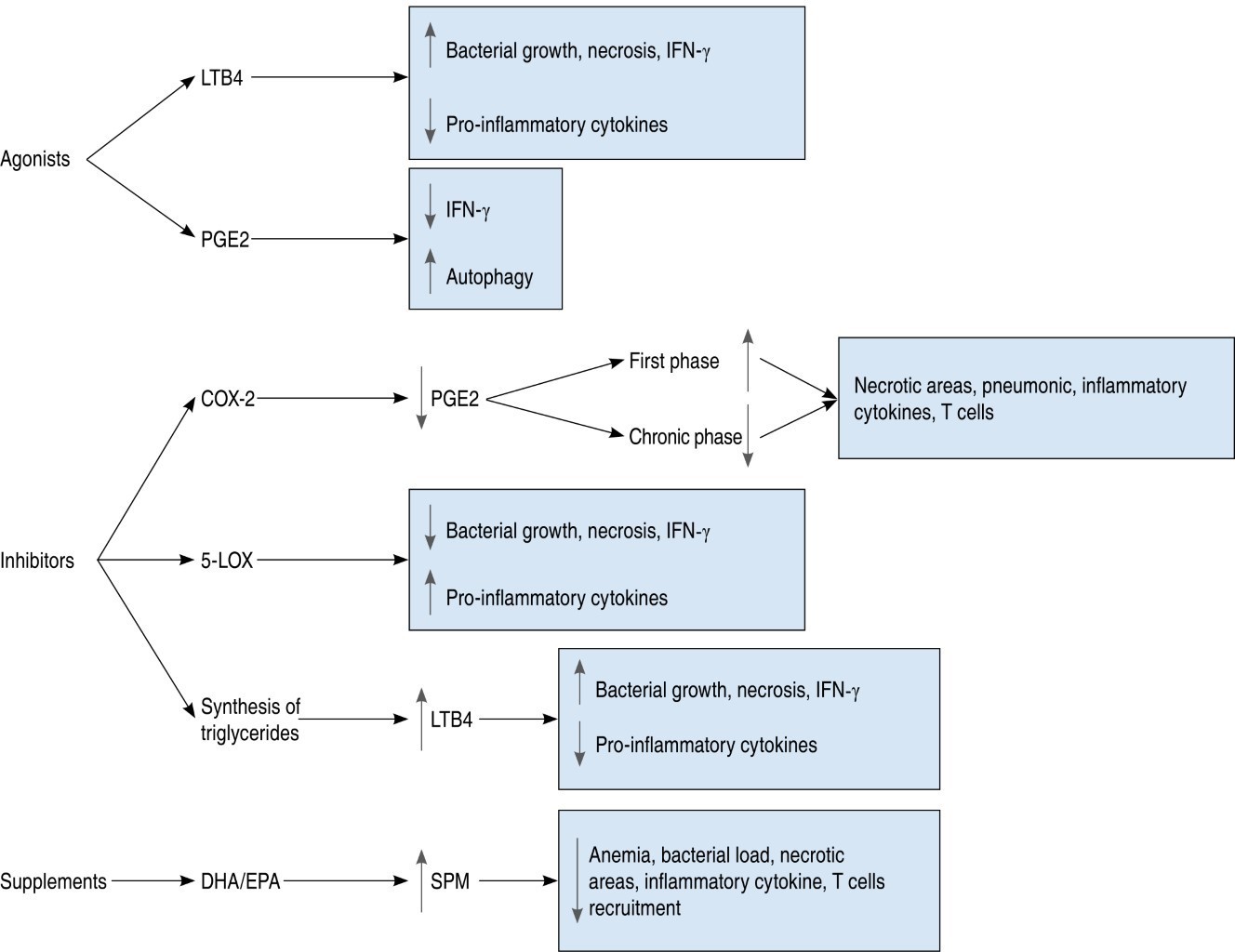
Post-infection interventions have allowed us to understand the scope of these host-directed therapies (Figure 2). Currently, three metabolic pathways involved in eicosanoid synthesis have been inhibited: COX-2, 5-LOX and triglyceride synthesis. COX-2 inhibitors decrease PGE2 production causing different effects depending on the time at which they are administered;43 at the onset of the disease, they induce an increase in necrotic areas, a greater amount of inflammatory cytokines and T cells;37 on the other hand, COX-2 inhibition in chronic stages decreases lesions and disease severity.36 In general, no interference with the use of conventional antibiotics was found.
Inhibition of 5-LOX reduces bacterial growth and necrosis, but increases pro-inflammatory cytokines;23 it is unknown whether this effect is due to increased LXA4 or reduced LTB4. Triglyceride synthesis up-regulates 5-LOX, but inhibition of triglycerides reduces bacterial load, possibly because it represents nutrient depletion.38,40 With the use of DHA/EPA dietary supplements, animal models reveal a reduction in anemia, inflammation and bacterial load and increased SPM synthesis.54 The impact this intervention would have in humans is unknown; however, SPM derived of DHA/EPA, MAR1 and RvD1 in in vitro cultures of human cells are known to have cell regeneration and inflammation-lowering effects.25 However, studies on genetic polymorphisms report an increased susceptibility to disease in populations with variations in 5-LOX,8 LTA4H55 or EP256 genes; while others report no associations between these same genes and disease severity.57-59
Although animal models offer a variety of resources for the study of tuberculosis, some limitations of currently used experimental models prevent critical analysis and implementation of therapeutic interventions in humans. For example, not all mouse strains experience a predominantly inflammatory response, whereas immunocompetent humans experience exacerbation of inflammation.35 The variety of mouse strains used influences the outcome; in many cases it was necessary to modify some gene to allow the study of the metabolic pathway of interest.60 C3HeB/FeJ and Sv129 mice are extremely susceptible to M. tuberculosis infection, and C57BL/6J and BALB/c mice are resistant.61-63 In mice, the biological action of PGE2 is mediated by four prostanoid receptor-linked proteins EP1, EP2, EP3 and EP4. These receptors are also expressed in human macrophages.14 However, in infected murine macrophages there is a higher amount of EP4 compared to EP2, which does not occur in human macrophages.64
In humans, the most frequent form is pulmonary tuberculosis; however, in research, mainly plasma and blood cells from donors are studied, which do not reflect what occurs in the alveolar space. Currently, different types of CT scans have been used to monitor the natural history of the disease, but it is difficult to carry out experimental studies in humans.65
Other animal models have been used. For example, rabbits infected with M. tuberculosis HN878 produce lesions similar to those found in humans. The distribution patterns of AA within the granuloma are similar in humans and rabbits,15 which would make the rabbit a better model; however, in rabbits M. bovis strains are normally used for the study of tuberculosis,65 rabbits are more expensive to maintain and their high susceptibility to stress demands strict control of environmental factors. In addition, the different biological responses between breeds and the positions of the various animal advocacy associations preclude further information.66
Regarding supplementation with ω-3 (omega-3), the experimental models are very diverse and do not predict the expected result in humans. Even the experimental doses used do not realistically represent the dietary intake in humans;67 the Food and Agriculture Organization of the United Nations (FAO) recommends a daily intake of 250 mg of EPA + DHA.68 In recent years, the demand for supplements has been increasing, but the necessary amount of consumption of each of them separately is unknown, since each one has a different metabolism.69 In Mexico, the average consumption of DHA/EPA is also unknown; however, following the COVID-19 pandemic, the consumption of fish (main source of these fatty acids) was reduced in households by 27-43%.70 For laboratory animals, the latest National Research Council (NRC) nutritional requirement tables published in 1995 do not specify the amounts of PUFA needed in the diet,71 but it is known that their administration is important to avoid a fatty acid deficiency that causes signs such as dermatitis, fatty liver, weight loss and reproductive problems.72 Dietary recommendations are changing according to new discoveries in the nutritional area.68,69
Finally, ex vivo/in vitro studies do not fully reflect the complexity of lung structure and pathogen-host interactions.73 Ex vivo whole blood studies have the advantage over in vitro cultures in that they allow us to evaluate the integration of the effects of antimycobacterial therapies through the host immune response37 and allow us to get closer and closer to understanding the molecular mechanisms involved in TB.
Requirements for future experimental designs
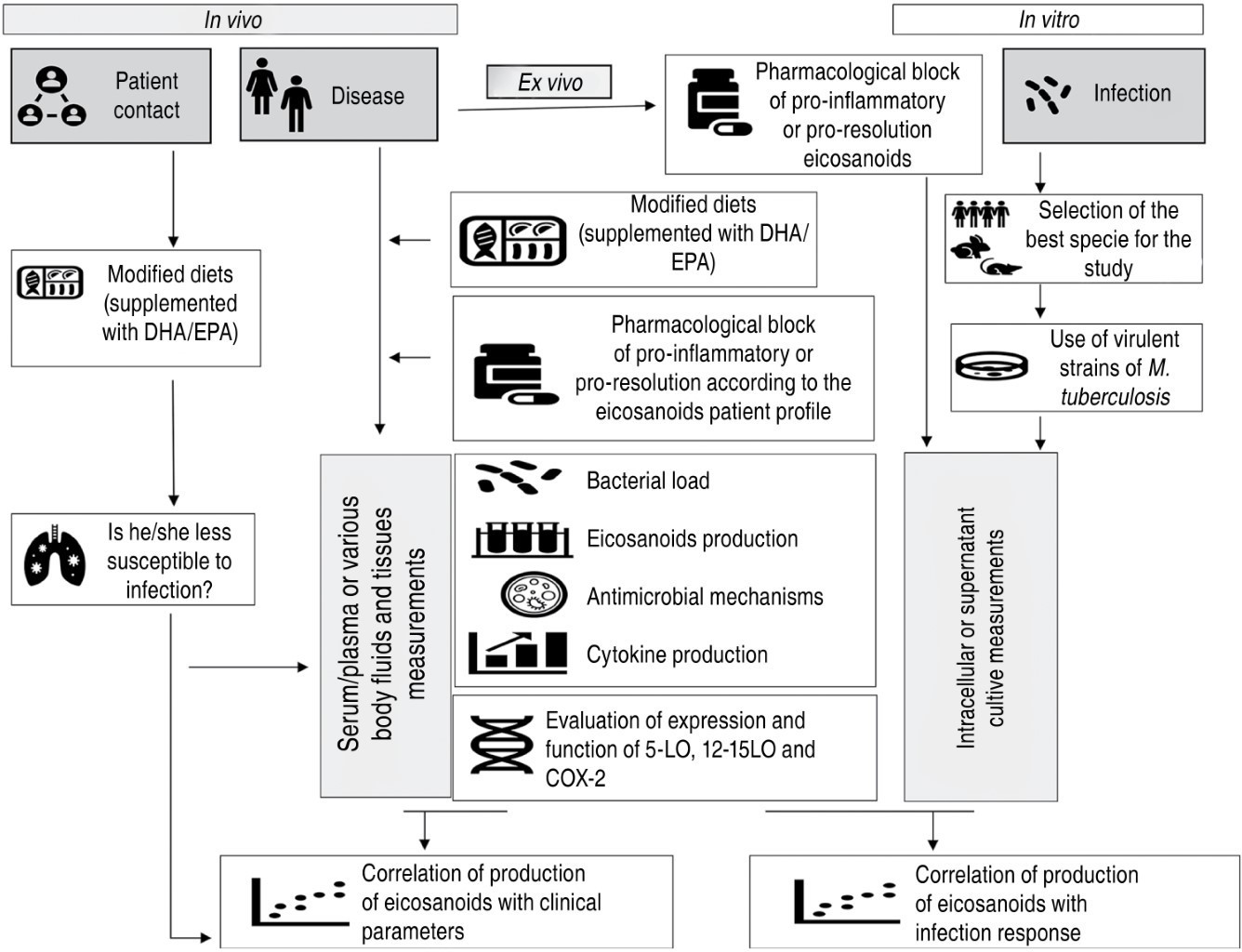
Interventions in the metabolic pathway of eicosanoids offer different therapeutic targets for TB that allow reducing pulmonary inflammation to preserve lung functionality without loss of antimicrobial immunity. For future research to better understand the mechanism of action of eicosanoids and to propose effective therapeutic schemes in TB, experimental strategies need to be optimized (Figure 3). Whether in vivo, in vitro and ex vivo investigations, the use of virulent TB strains will be important to better understand host-parasite metabolic interactions. In addition, it is necessary to prioritize research in humans, in vitro and ex vivo, both TB patients and their contacts. To know the real scope of interventions in the eicosanoid pathway, it will be necessary to measure bacterial load, cytokine production and cellular antimicrobial activity, as well as to define the complete profile of eicosanoid production with a view to personalized medicine, taking into account the previous profile and the phase of the disease in which the patient is.
Conclusions
Because eicosanoids offer therapeutic targets of interest for TB, it is important to optimize experimental models and their impact on the generation of these targets. Diets with DHA/EPA supplementation and pharmacological blockade of either pro-inflammatory or pro-resolution eicosanoids could be beneficial to both the patient and the patient contact. Eicosanoids not only have roles in the inflammatory response, but also act as mediators of the pathogenesis process, so further research is needed to better understand the potential of eicosanoids as future host-directed therapies.
Acknowledgements
Esmeralda Juárez is grateful for the support of the CONACyT (grant A3-S-35173).
AFILIACIONES
1Universidad Nacional Autónoma de México, Mexico City, Mexico 2Instituto Nacional de Enfermedades Respiratorias Ismael Cosío Villegas, Mexico City, Mexico.Conflict of interests: the authors declare that they have no conflict of interests.
REFERENCES
Guzmán-Beltrán S, Carreto-Binaghi LE, Carranza C, Torres M, Gonzalez Y, Muñoz-Torrico M, et al. Oxidative stress and inflammatory mediators in exhaled breath condensate of patients with pulmonary tuberculosis. A pilot study with a biomarker perspective. Antioxidants (Basel). 2021;10(10):1572. doi: 10.3390/antiox10101572.
Vinhaes CL, Oliveira-de-Souza D, Silveira-Mattos PS, Nogueira B, Shi R, Wei W, et al. Changes in inflammatory protein and lipid mediator profiles persist after antitubercular treatment of pulmonary and extrapulmonary tuberculosis: A prospective cohort study. Cytokine. 2019;123:154759. doi: 10.1016/j.cyto.2019.154759.
Sorgi CA, Soares EM, Rosada RS, Bitencourt CS, Zoccal KF, Pereira PAT, et al. Eicosanoid pathway on host resistance and inflammation during Mycobacterium tuberculosis infection is comprised by LTB4 reduction but not PGE2 increment. Biochim Biophys Acta Mol Basis Dis. 2020;1866(3):165574. doi: 10.1016/j.bbadis.2019.165574.
Liu S, Xie Y, Luo W, Dou Y, Xiong H, Xiao Z, et al. PE_PGRS31-S100A9 Interaction Promotes Mycobacterial Survival in Macrophages Through the Regulation of NF-κB-TNF-α Signaling and Arachidonic Acid Metabolism. Front Microbiol. 2020;11:845. Available in: https://www.frontiersin.org/article/10.3389/fmicb.2020.00845/full
Peres-Buzalaf C, de Paula L, Frantz FG, Soares EM, Medeiros AI, Peters-Golden M, et al. Control of experimental pulmonary tuberculosis depends more on immunostimulatory leukotrienes than on the absence of immunosuppressive prostaglandins. Prostaglandins Leukot Essent Fatty Acids. 2011;85(2):75-81. doi: 10.1016/j.plefa.2011.04.024.
Rangel Moreno J, Estrada García I, De La Luz García Hernández M, Aguilar Leon D, Marquez R, Hernández Pando R. The role of prostaglandin E2 in the immunopathogenesis of experimental pulmonary tuberculosis. Immunology. 2002;106(2):257-266. Available in: http://doi.wiley.com/10.1046/j.1365-2567.2002.01403.x
Hernández-Pando R, Orozco-Esteves H, Maldonado HA, Aguilar-León D, Vilchis-Landeros MM, Mata-Espinosa DA, et al. A combination of a transforming growth factor-β antagonist and an inhibitor of cyclooxygenase is an effective treatment for murine pulmonary tuberculosis. Clin Exp Immunol. 2006;144(2):264-272. doi: 10.1111/j.1365-2249.2006.03049.x.
Narendran G, Kavitha D, Karunaianantham R, Gil-Santana L, Almeida-Junior JL, Reddy SD, et al. Role of LTA4H polymorphism in tuberculosis-associated immune reconstitution inflammatory syndrome occurrence and clinical severity in patients infected with HIV. PLoS One. 2016;11(9):e0163298. doi: 10.1371/journal.pone.0163298.
Reeves PG, Nielsen FH, Fahey GC. AIN-93 purified diets for laboratory rodents: final report of the American Institute of Nutrition ad hoc writing committee on the reformulation of the AIN-76A rodent diet. J Nutr. 1993;123(11):1939-1951. Available in: https://academic.oup.com/jn/article/123/11/1939/4722783
Jontvedt Jorgensen M, Nore KG, Aass HCD, Layre E, Nigou J, Mortensen R, et al. Plasma LOX-products and monocyte signaling is reduced by adjunctive cyclooxygenase-2 inhibitor in a phase i clinical trial of tuberculosis patients. Front Cell Infect Microbiol. 2021;11:669623. doi: 10.3389/fcimb.2021.669623.