Introduction
COVID-19, associated with SARS-CoV-2, has caused millions of deaths worldwide. Unfortunately, the pandemic that emerged in 2019 in the Chinese province of Wuhan has spread for several years and new variants of the virus have appeared, such as Omicron and some of its variants. At the beginning of the pandemic, in February 2020, at the National Institute of Respiratory Diseases Ismael Cosío Villegas (INER), one of the first confirmed cases for this virus in Mexico was diagnosed, although very likely there were other cases that may have coexisted with this index case in southern Mexico City. A wise decision by the authorities and institutions, such as INER and other national health institutes in Mexico, was to anticipate and be prepared in response to reports of this outbreak that rapidly spread through China and other countries, and an alert that was issued in the final weeks of 2019 by the authorities of the Mexican Ministry of Health. This allowed the establishment of operational strategies for diagnosis and management of respiratory disease cases by coronavirus and the reestablishment of reference and sample confirmation systems through molecular biology techniques. At that time, INER, by order of the authorities, was converted into a hospital specifically dedicated to the diagnosis and management of severe COVID-19 cases. However, this did not occur in parallel at other levels of care. The lack of early diagnosis and confinement of new cases and their contacts at that time were factors associated with higher morbidity and mortality. Even now, preventing the spread of SARS-CoV-2 remains a public health challenge. The lack of access to specific antivirals and vaccines has also contributed significantly to the development of severe forms and poor clinical outcomes in COVID-19 patients.
The extensive research and literature on the characteristics of SARS-CoV-2, its origin, its mechanisms of infection, replication and cellular tropism, as well as on host characteristics and risk factors for infection and development of severe forms of the disease, has allowed the development of a series of prophylactic, therapeutic, and infection control strategies with a speed never before seen in the history of applied medicine. Despite this, understanding of the viral and host factors that determine the clinical behavior of the disease is still incomplete. Through advanced genome expression analyses, molecular signatures associated with severe disease have been identified that are currently used as prognostic biomarkers to guide therapeutic decisions1–8.
The mechanisms of immune response have been identified, both the innate response of cells such as macrophages and the adaptive response of T and B lymphocytes that determine poorly regulated inflammatory responses that can contribute to tissue damage. An example of this is the histopathological evidence of hemophagocytosis in bone marrow and reticuloendothelial organs, and the macrophage activation syndrome observed in virus-induced hemophagocytic lymphohistiocytosis, which suggests that the host’s innate immune system plays a crucial role in the immunopathology of COVID-19. In fact, several studies have highlighted the efficacy of some immunomodulatory agents that allow attenuation of the inflammatory response to SARS-CoV-2 and prevent lung injury.
It is important to note that an increasing incidence of sequelae was observed in patients recovering from COVID-19, including pulmonary fibrosis, especially among those who recovered from severe disease. These complications could permanently affect the respiratory function of patients, negatively impacting their quality of life. Therefore, it is a priority to study the pathogenic processes underlying excessive inflammation, tissue injury, and tissue remodeling mechanisms, including the extracellular matrix, after SARS-CoV-2 infection.
In this review, we present an overview of the mechanisms of SARS-CoV-2 infection and the immune processes of protection and damage associated with different clinical forms of COVID-19.
Biology of SARS-CoV-2
SARS-CoV-2 is a member of the human coronavirus (HCoV) group, consisting of HCoV-229E, HCoV-NL63, HCoV-HKU1, HCoV-OC43, SARS-CoV-1, and MERS-CoV1. These pathogens are single-stranded RNA coronaviruses belonging to the Coronaviridae family. Some of them have caused a variety of respiratory diseases of varying severity in the past; for example, SARS-CoV-1 infected more than 8,000 people in Asia in 20032, and MERS-CoV (Middle East respiratory syndrome coronavirus) originating in Saudi Arabia in 2012 had high mortality rates3,4.
Through comparative studies of viral genome sequences, HCoVs can be grouped into four genera: alpha, beta, gamma, and delta. The novel SARS-CoV-2 is a Betacoronavirus genetically related to a bat coronavirus called BatCoV RaTG13, as well as to SARS-CoV-15,6. Furthermore, SARS-CoV-2 shares genetic identity with some coronavirus isolated from pangolins7,8. Therefore, COVID-19 is believed to be a zoonotic disease originating in bats with the pangolin as a possible intermediate host. The SARS-CoV-2 genome consists of an RNA strand of 29,903 base pairs that encodes a replicase-transcriptase and the structural proteins spike (S), envelope (E), membrane (M), and nucleocapsid (N)5.
Mechanism of SARS-CoV-2 infection
The initial step in the SARS-CoV-2 infection process is the recognition of its receptor on the membrane of host cells. This process is mediated by the S protein, which recognizes human angiotensin-converting enzyme 2 (ACE2), the same receptor for the SARS-CoV-1 S protein9–11. This protein has two functional domains: the S1 domain contains the receptor binding domain (RBD) that binds to ACE2, while the S2 domain performs fusion of the viral membrane with the target cell membrane11. Therefore, the distribution of the ACE2 receptor in different organs and tissues is crucial for determining the infectivity and tropism of the virus.
A key aspect of the infection process is the activation of the S protein. This process is mediated by different host proteases that cleave the S protein at its S1/S2 and S’2 sites. This protein processing allows full functional activity of the S2 domain of the S protein, so it can fuse the viral and cellular membranes. For this purpose, just as SARS-CoV-1 does, SARS-CoV-2 employs transmembrane protease serine 2 (TMPRSS2)9,12,13. Interestingly, the proteases TMPRSS4 and cathepsin L14,15, as well as the human CD147 receptor16, also promote SARS-CoV-2 infection. Therefore, the tissue expression pattern of these elements may determine viral tropism, and even some drugs that inhibit the activity of these proteases or the CD147 receptor have been proposed as therapeutic agents to prevent and treat COVID-199,15,16.
Another factor involved in the SARS-CoV-2 infection process is phosphatidylinositol-3-phosphate kinase (PIKfyve, phosphoinositide kinase, FYVE-type)15. This enzyme regulates the production of phosphatidylinositol-3,5-bisphosphate, a phospholipid that participates in the endosome maturation process. In fact, apilimod, a potent PIKfyve inhibitor, reduces SARS-CoV-2 infectivity and could be a candidate for therapeutic purposes15.
Once the virus manages to enter cells, viral replication begins with translation of the replicase-polymerase gene and assembly of the replication-transcription complex. This complex transcribes the genomic regions of the virus that encode structural proteins. In this way, new virions are produced in the endoplasmic reticulum and Golgi apparatus and are then released from the cell1. A particular characteristic of SARS-CoV-2 is that it possesses a polybasic furin cleavage sequence at the S1/S2 site, absent in other related coronaviruses6,11. This sequence is processed in the Golgi apparatus during the biosynthesis of the S protein of new virions11. Therefore, new SARS-CoV-2 virions possess an activated S protein ready to bind to the ACE2 receptor, without requiring the activity of other host proteases. Since virtually all human cells express furin, the insertion of a furin cleavage sequence could increase the transmission capacity and tropism of SARS-CoV-2.
Immune response against SARS-CoV-2
It is known that once SARS-CoV-2 achieves its first phase of replication in the upper respiratory tract, it spreads early to the lower respiratory tract, where it triggers a pronounced innate immune response that, in symptomatic cases, leads to the onset of clinical manifestations. Among the most frequent signs and symptoms observed in people with COVID-19 are fever, dry cough, fatigue, headache, diarrhea, dyspnea, anosmia, and loss of taste17,18. However, the disease has a heterogeneous clinical spectrum that includes asymptomatic cases, patients with mild manifestations, and patients with moderate to severe symptoms who develop acute respiratory failure, multiorgan dysfunction, and risk of fatality19,20. Fortunately, 85% of people infected with SARS-CoV-2 do not present clinical manifestations or suffer mild disease, while only 5-30% of cases may present a critical form21-23. This means that, in the vast majority of COVID-19 cases, the immune system is effective in controlling the infection and eliminating the virus. However, as already mentioned, the immune components that participate in protective responses against SARS-CoV-2 are still under study.
Traditionally, it is believed that defense against viral infection involves activation of mechanisms such as the production of type I interferons, which limit pathogen replication within infected cells and prevent its spread to healthy cells. Likewise, a robust humoral response with production of high-affinity antigen-specific antibodies is required to neutralize viral particles. Finally, cellular immunity plays a key role in identifying and lysing infected cells to eliminate intracellular reservoirs of the pathogen. All these mechanisms may be important for protection against COVID-19: the current evidence on the role of different immune factors during SARS-CoV-2 infection is summarized below.
Innate immune response
The innate immune system provides nonspecific protection against a wide variety of microorganisms. Its main components include epithelial and mucosal barriers, as well as humoral and cellular factors that together can recognize pathogens and initiate an inflammatory response that, in most cases, is sufficient to eliminate the infection. Mucosal barriers, such as the respiratory epithelium, have a series of mechanisms that prevent the adhesion of pathogens to the surface of epithelial cells and the initiation of infection. The respiratory mucosa is important because any alteration of its integrity, as in the case of people with chronic bronchial and pulmonary diseases, confers a greater risk of contracting COVID-19 and presenting a severe form of the disease.
Among the barrier mechanisms that protect the host is the production of mucus in the airways, which traps and allows the elimination of viruses and bacteria, as well as the production of surfactant in the alveolar epithelium, which contains some protein defense factors that allow neutralization of different microorganisms. Such is the case of surfactant protein D (SP-D), which can bind to the S protein of SARS-CoV-2 and neutralize it, inhibiting virus entry into epithelial cells24. However, some studies suggest that, in severe cases of the disease, the virus is able to counteract the effects of surfactant by suppressing SP-D production25,26. Other molecular elements produced by the respiratory epithelium that are suppressed during infection include some cytokines with antimicrobial properties, such as the chemokine CXCL1727.
Once SARS-CoV-2 overcomes the barrier mechanisms, the next step in defense against the virus includes recognition of pathogen-associated molecular patterns, such as the viral single-stranded RNA or envelope proteins, by surface or intracellular pattern recognition receptors expressed on epithelial cells, alveolar macrophages, and other cells of the phagocytic-mononuclear system. Currently, not all innate immune receptors that recognize viral infection and initiate immune responses against SARS-CoV-2 are known. Since this virus is genetically related to SARS-CoV-1, it is presumed that both share infection mechanisms. In this sense, SARS-CoV-1 is recognized by Toll-like receptors (TLR) TLR3 and TLR4, which induce an immune reaction through the MyD88 and TRIF pathways28,29. Furthermore, SARS-CoV-1 triggers the production of interleukin (IL)-1β through inflammasome activation30, with the aim of triggering an inflammatory response against the virus. In this regard, recent research indicates that TLR2 is another innate sensor capable of recognizing the virus through its binding to the envelope protein. Once this recognition occurs, TLR2 induces an inflammatory reaction through the MyD88-dependent signaling pathway31. Inflammasome activation is also likely to occur during SARS-CoV-2 infection, as high levels of IL-1β have been observed in patients with COVID-19, as described below32. Studies have shown that SARS-CoV-2 infection induces activation of NLRP3 inflammasomes and release of IL-1β in monocyte cultures and peripheral blood mononuclear cells from COVID-19 patients; however, excessive inflammasome activation can also lead to the development of severe forms of the disease33,34.
Another crucial innate mechanism in defense against viral infections is the production of type I interferons (IFN) (primarily IFN-α and IFN-β), which bind to membrane surface receptor complexes known as the IFN-α/β receptor (IFNAR), consisting of IFNAR1 and IFNAR2 chains. Once bound to their receptor, type I IFNs can interfere with virus replication in host cells, activating different signaling pathways involving antiviral proteins such as PKR. In vitro studies indicate that type I IFN activity is effective in inhibiting SARS-CoV-2 replication in human cells35. In primate COVID-19 models, it has been observed that the IFN-mediated response is widely induced in the lungs and is crucial for defense against infection36. However, it is possible that suppression of type I IFN is a SARS-CoV-2 strategy to evade the innate antiviral immune response, as some studies have shown very low production of IFN-α and IFN-β in the plasma of Mexican and French patients with severe forms of COVID-1937,38.
Finally, some cells of the innate immune system could play an important role in host defense. Among these cells are alveolar macrophages, which are presumed to be the first immune system cells to come into contact with SARS-CoV-2. Although these cells can recognize the virus, their response is manipulated by the pathogen39,40. Other crucial cells in defense against viral infections are NK (natural killer) cells, but their role in COVID-19 is not yet fully understood. In general, most studies characterizing the immunological profile of COVID-19 patients show that NK cells are decreased in the blood of those with severe disease41,42. It is believed that some deficiencies in the expression of NK cell receptors, such as NKG2C, could be associated with a higher risk of severe infection43. This suggests a possible protective role of NK cells in the immune response against SARS-CoV-2.
Adaptive immune response
The adaptive immune system provides a second line of defense against pathogens mediated by lymphocytes that possess antigen-specific receptors capable of undergoing genetic recombination. Therefore, adaptive immunity against viruses is capable of controlling infection effectively in most cases, through mechanisms specifically directed at the attacking pathogen. Adaptive immunity cells can promote protective responses against pathogens through the production of proinflammatory and antiviral cytokines by CD4+ helper T lymphocytes, or through the destruction of infected cells mediated by CD8+ cytotoxic T lymphocytes. These cells are believed to play a fundamental role in defense against SARS-CoV-2, as it has been observed that in other coronavirus infections, antigen-specific T lymphocytes are crucial for conferring protection. In fact, in people who were affected by SARS-CoV-1 in 2003 and survived the disease, it has been proven that different virus-specific memory T lymphocyte clones survive and are capable of responding to the virus up to 17 years after the primary infection44. Some studies suggest that individuals infected with SARS-CoV-2 develop CD4+ and CD8+ memory T lymphocytes, which persist in circulation for up to 8 months45. These memory responses also include follicular T lymphocytes, which are crucial for supporting B lymphocytes in the production of protective antibodies. Despite the above, current data on the role of cellular immunity in COVID-19 pathogenesis are still unclear and controversial. For example, some researchers have found that patients with severe forms of the disease have reduced numbers of T lymphocytes in circulation, but the antigen-specific T lymphocytes of these individuals develop more potent cytokine production responses46.
The humoral response is also important for controlling viral infections in humans. In this sense, B cells from individuals infected with SARS-CoV-2 undergo immunoglobulin (Ig) class switching from IgM to higher affinity IgG and IgA. Thus, sera from patients recovering from COVID-19 contain anti-S protein IgG, IgM, and IgA antibodies and anti-nucleocapsid IgG antibodies. However, elevated titers of IgG and IgM antibodies have been found in patients with severe disease, which questions the protective capacity of antibody responses in COVID-1947. It is important to address the neutralizing capacity of antibodies induced by natural infection and vaccination, especially to improve the protective capacity of the humoral response elicited by vaccines. A study by Dan et al.45 reveals that even when there are different kinetics of neutralizing antibodies against different components of SARS-CoV-2, antigen-specific memory B cells remain detectable for more than 8 months after symptom onset in some COVID-19 patients. Ellebedy et al. have observed different clones of long-lived plasma cells and memory B lymphocytes in the bone marrow and blood of people who recovered from COVID-19 up to 11 months after infection. These plasma cells and memory B lymphocytes can respond to secondary encounters with SARS-CoV-2 and produce neutralizing antibodies48. Finally, the importance of adaptive immune memory responses mediated by B lymphocytes for defense against COVID-19 is also highlighted in studies that have shown that the germinal center reaction in lymph nodes is abolished or altered in severely ill people after SARS-CoV-2 infection49.
The defense mechanisms against SARS-CoV-2 are summarized in figure 1.
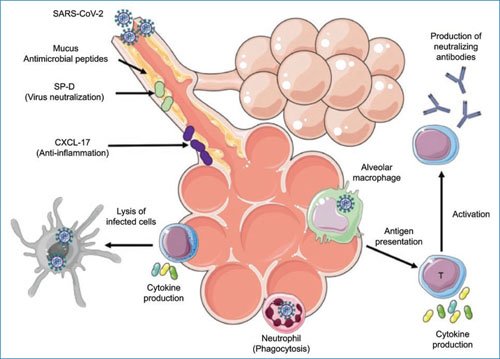
Figure 1. Defense mechanisms of immune immunity against SARS-CoV-2. NK: natural killer cell; SP-D, surfactant protein D.
Immunopathology of severe forms of COVID-19
Cytokine storm and immunosuppression
People who progress to severe COVID-19 develop pneumonia within 10-20 days after symptom onset, which is associated with reduced oxygen saturation, acute respiratory distress syndrome, and prominent lung damage with ground-glass opacities. Acute respiratory distress syndrome is characterized by increased pulmonary permeability, severe hypoxemia, and non-cardiogenic pulmonary edema. These conditions alter the alveolar-capillary barrier and are a consequence of a systemic hyperinflammation process50–52.
For guiding immunotherapeutic interventions in critically ill patients, a better understanding of host factors involved in protective versus pathogenic immunity against SARS-CoV-2 is crucial. Unfortunately, what we understand today about the immunopathology of severe COVID-19 is a paradox: the adaptive response is hyperactive but unable to control the virus. In fact, COVID-19 patients present a profile of proinflammatory cytokines (IL-1β, IL-2, IL-6, IL-7, IL-8, IL-9, FGF, G-CSF, GM-CSF, IFN-γ, CXCL10, CCL2, CCL3, CCL4, PDGF, TNF-α, and VEGF) and regulatory cytokines (IL-10 and TGF-β)21,53–55,53-55. This phenomenon has been termed “cytokine storm” and mediates tissue damage in COVID-19 patients who progress to severe disease55,58. Of these factors, IL-1β, IL-6, CXCL10, and TNF-α are the cytokines most strongly associated with tissue damage in different organs, including the brain, due to their inflammatory properties. For example, IL-1β and IL-6 have been implicated in neurotoxicity associated with chimeric antigen receptor T cell therapy in patients with hematologic neoplasms59,60. These cytokines possess detrimental effects on endothelial function in various vascular niches, which may be implicated in the pathophysiology of thrombotic and neurological complications of COVID-19. In this regard, it is important to mention that other organs, in addition to the lungs, can also be affected by SARS-CoV-2, such as the brain, heart, intestines, kidneys, and liver, among others.
This generalized inflammatory reaction also induces the production of several acute phase reactants, such as C-reactive protein and ferritin in the liver, which further increase the release of inflammatory mediators and enrich cytokine release in patients with severe COVID-1961,62. In fact, elevated ferritin levels predict the risk of death in these patients62. Furthermore, the hyperinflammation observed in critically ill COVID-19 patients may be associated with the development of thrombotic events. In this sense, abnormal coagulation parameters, such as higher D-dimer levels and longer prothrombin and activated partial thromboplastin times, have been associated with unfavorable prognosis. These abnormal coagulation parameters occur shortly after hospitalization, and in some patients, fibrinogen concentrations and antithrombin activity decrease over time63–65.
Interestingly, unlike other cytokine storm syndromes, the polyfunctional immune activation of COVID-19 is accompanied by lymphocytopenia and strong infiltration of immune cells, primarily mononuclear (lymphocytes and macrophages), into the lung interstitium66–68. In patients with severe SARS-CoV-2 infection, a wide range of immune cell subtypes is decreased in circulation. These cells include monocytes, dendritic cells, CD4+ and CD8+ T cells, and NK cells41. Furthermore, the few adaptive lymphocytes remaining in the blood express markers of functional exhaustion69. These data suggest that severe COVID-19 produces an immunosuppression state similar to that induced by sepsis70; it is also possible that robust recruitment of functional immune cells to SARS-CoV-2 infection sites may explain the leukocytopenia observed during COVID-19. A recent study by Remy et al.71 has demonstrated that immunosuppression observed in COVID-19 is even more profound than that of critically ill patients with sepsis from other causes. These researchers observed that IFN-γ production by peripheral blood T cells from COVID-19 patients was impaired compared to T cells from healthy individuals and sepsis patients after stimulation with anti-CD3/anti-CD28 antibodies; reduced TNF-α production was found by stimulated monocytes from COVID-19 patients. These findings led the researchers to propose that the primary immune mechanism underlying COVID-19 morbidity and mortality is immunosuppression rather than hyperinflammation.
The immune profile in severe COVID-19 shows distinct features compared to other respiratory infections, such as pandemic influenza A (H1N1). Among the immune factors found only in critically ill COVID-19 patients, and not in influenza patients, are IFN-γ, IL-4, IL-5, IL-6, IL-10, IL-12, IL-13, IL-1β, CCL11, VEGF, TWEAK, TSLP, MMP-1, and MMP-338. These molecules could play a specific role in COVID-19 and are potential targets for reducing its morbidity and mortality. It is noteworthy that higher levels of Th2 cytokines, particularly IL-4 and IL-5, could inhibit protective Th1 antiviral responses in COVID-19 patients. Therefore, the lack of immune balance in the type of effector response is another crucial determinant of the collapse of protective immunity in the host against SARS-CoV-2. This Th2-skewed response can generate interstitial infiltrates of Th2 cells, neutrophils, eosinophils, and type 2 innate lymphoid cells, which mediate pulmonary inflammation and tissue damage. Critically ill COVID-19 patients often show interstitial pulmonary infiltrates, some of which resemble various forms of progressive interstitial lung disease, such as cryptogenic organizing pneumonia and nonspecific interstitial pneumonia21,72–74. These harmful effects of Th2 responses could also explain the abnormalities in lung function and progression to pulmonary fibrosis observed in more than 45% of COVID-19 patients discharged from hospitals, particularly elderly subjects. Therefore, it would be of great interest to characterize the cytokine profile of COVID-19 patients who subsequently develop any form of interstitial lung disease, as they would benefit from specific antifibrotic therapies.
Neutrophil infiltration and lung damage
A variety of inflammatory mediators released excessively during severe SARS-CoV-2 infection function to mediate the recruitment of myeloid cells to sites of inflammation. One of the cells that heavily infiltrate the lungs of patients with severe COVID-19 is neutrophils, probably following a chemotactic gradient generated by high concentrations of IL-8, CXCL10, CCL2, and CCL3 released into circulation21,53–55. Neutrophils are the most abundant leukocytes in blood and have a short half-life. Due to their mobility, they can easily infiltrate inflamed tissues and contribute to the defense response against different microorganisms. Their main function is to mediate phagocytosis of pathogens, as well as release cytokines and proteolytic substances contained in their granules. Neutrophils can exert a protective role in defense against viral infections; it is well known that neutrophils are always present in the lungs of patients with different respiratory infections associated with the development of acute respiratory distress syndrome, especially due to their capacity for degranulation and lysis, promoting tissue damage.
In line with these findings, in animal models of macaques, it has been observed that neutrophils infiltrate the lung rapidly during SARS-CoV-2 infection36. This is also observed in COVID-19 patients, in whom it has been found that neutrophilia and a higher neutrophil ratio is a distinctive characteristic of severe cases and those occurring in elderly patients, associated with unfavorable prognosis36,77. In patients who died from severe COVID-19, intense neutrophil infiltration has been observed in pulmonary capillaries, extravasation into alveolar spaces, and neutrophilic mucositis in the airways78. Within sites of SARS-CoV-2 infection, neutrophil degranulation results in the release of proteases capable of amplifying the inflammatory response. For instance, cathepsin G stimulates increased production of cytokines and chemokines, promoting recruitment of monocytes, macrophages, and neutrophils, as well as increased endothelial and epithelial permeability36,79. Cathepsin G also activates some matrix metalloproteinases (MMP), initiating the alteration of mechanisms that regulate the composition of the extracellular matrix (ECM) and are associated with the appearance of sequelae characteristic of post-COVID-19 syndrome, as described below. Finally, neutrophils are a source of excess extracellular traps, which further exacerbate the cytokine storm and perpetuate a vicious cycle of inflammation and neutrophil recruitment to the lung. Therefore, the search for new strategies to reduce COVID-19 morbidity and mortality focusing on interrupting neutrophil recruitment to the lung and limiting lung damage induced by the contents of these cells’ granules is justified.
Immune mechanisms associated with post-COVID-19 syndrome
Post-COVID-19 syndrome or post-acute sequelae of COVID-19 (PASC) is defined by disease persistence for more than 28 days after symptom onset. Various longitudinal studies suggest that this process can be observed in 30% to 80% of individuals who suffer SARS-CoV-2 infection. In this regard, a notable characteristic of the COVID-19 convalescent phase is that many patients who suffered severe lung damage remain with permanent pulmonary dysfunction. Many of the chronic deleterious effects of COVID-19 are related to pulmonary fibrosis following injury. The mechanisms underlying the development of these complications have not been well defined until now. In patients with severe pneumonia of other etiologies, severe epithelial and endothelial damage accompanied by extensive fibrosis has frequently been observed. Patients presenting greater fibrotic changes are those who required prolonged periods of mechanical ventilation (~12 days) and developed more severe systemic organ failure50,81. Interestingly, pulmonary fibrosis is also a long-term sequela in patients with pandemic influenza A (H1N1). Several mechanisms, including barotrauma associated with mechanical ventilation, oxygen toxicity, and hyperinflammation, are crucial in determining sequelae in these patients after recovery from severe disease82. These factors cause mild epithelial lesions that are not adequately repaired, leading to fibroblast hyperactivation, excessive ECM deposition, and pulmonary parenchyma remodeling83.
It is not understood to what extent mechanical injury and hyperinflammation contribute to pulmonary fibrosis in COVID-19. A significant number of severely ill COVID-19 patients with acute respiratory distress syndrome require critical care and respiratory assistance, and those who survive show persistent ground-glass opacities, chronic pulmonary dysfunction, and pulmonary fibrosis that affect their quality of life84–86. On the other hand, hyperinflammation in severe COVID-19 also includes the release of inflammatory chemokines such as CCL2, CCL3, and IP-10. These chemotactic factors are associated with dysregulated activation of cells in the mononuclear phagocyte compartment, which could further promote hyperinflammation in COVID-19 patients. In fact, bronchoalveolar fluid from severe COVID-19 patients contains high concentrations of CCL2, CCL3, CCL4, and CCL7, and a decreased proportion of tissue-resident alveolar macrophages, but large amounts of monocyte-derived inflammatory macrophages87. Interestingly, this macrophage subpopulation expresses RNA transcripts that have been previously associated with tissue repair and promotion of fibrosis in liver cirrhosis88,89.
Surprisingly, during fibrosis, many types of collagen can modulate cellular functions and the physiological processes of leukocytes and parenchymal cells. Furthermore, in response to inflammation, ECM degradation by MMPs generates small peptides that can act as chemotactic factors for leukocytes, thereby increasing disease immunopathology. These mechanisms further promote MMP hyperactivity, causing progressive destruction of pulmonary parenchyma90. Therefore, MMPs and other ECM components could act as readouts of ongoing profibrotic activity and lung injury in severe COVID-19 patients, as discussed below.
An important controversy that has recently emerged is whether pulmonary fibrosis is an exclusive sequela of severe SARS-CoV-2 infection or whether patients with mild to moderate disease are also at risk. In this regard, evidence from recovering COVID-19 patients, both adult and pediatric, suggests that older patients and those with more severe disease develop fibrosis91–93. Similarly, a study conducted in Italy between March and April 2020 found that patients with non- severe manifestations showing pulmonary opacities showed complete remission of these lesions and no fibrosis during follow-up. In contrast, Dadhwal et al.94 reported five cases of patients with asymptomatic or mild symptomatic COVID-19 presenting dyspnea and chest images suggesting resolution of ground-glass opacities who subsequently developed fibrosis 4 to 8 weeks after diagnosis.
Despite all the studies conducted to understand the immunopathological bases of pulmonary and extrapulmonary chronic sequelae observed in patients with PASC or prolonged COVID-19, their etiology remains unclear. One of the essential characteristics of PASC is viral persistence in various tissues, such as lung, central nervous system, kidneys, and intestine95,96. It has been suggested that patients who develop PASC may have aberrant, poorly regulated immune responses, and overactivation of cell populations such as myeloid cells and T and B lymphocytes that infiltrate tissues, generating tissue scenarios rich in proinflammatory or profibrotic cytokines, as is the case of post-COVID-19 pulmonary fibrosis, characterized by elevation of TGF-β and hyperactivation of myofibroblasts producing extracellular matrix97.
In post-COVID-19 syndrome, persistent dyspnea, frequently accompanied by fatigue, chest discomfort, and cough, affects approximately 20% of patients 3 months after the initial SARS-CoV-2 infection98. Furthermore, a considerable number of patients, particularly those who overcome COVID-19 acute respiratory distress syndrome and require treatment with high-flow nasal oxygen or mechanical ventilation, present chronic pulmonary sequelae, as demonstrated by pulmonary function tests and radiological changes on chest computed tomography, such as ground glass and fibrosis99. Interestingly, some studies have shown that the use of antifibrotic drugs, such as pirfenidone and nintedanib, has positive clinical and functional effects for treating pulmonary sequelae of COVID-19100,101.
Post-viral pulmonary sequelae are not exclusive to SARS-CoV-2 and have been described after several other respiratory viral infections, possibly also associated with inflammatory alterations that lead to chronic pulmonary damage.
Taken together, these data highlight the need for further research studies in patients recovering from COVID-19 to establish better preventive, therapeutic, and rehabilitation strategies against pulmonary fibrosis. For these purposes, new biomarkers with predictive value will also be required to allow early detection of lung lesions and fibrosis.
The mechanisms involved in the pathophysiology of severe forms of COVID-19 and their sequelae are summarized in figure 2.
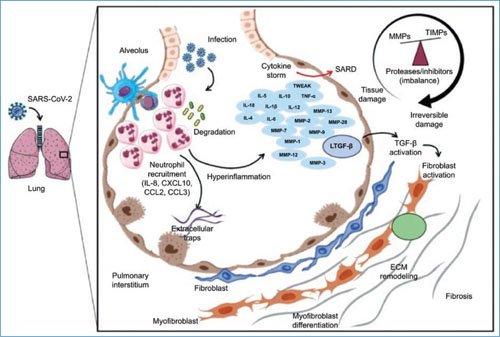
Figure 2. Immunopathology of severe forms of COVID-19 and their sequelae. ECM: extracellular matrix; MMPs: matrix metalloproteinases; ARDS: acute respiratory distress syndrome; TIMPs: tissue inhibitors of MMPs.
Conclusions
The spread of SARS-CoV-2 in various countries has caused possibly the greatest global health crisis of the last 100 years. Although the vast majority of COVID-19 cases present asymptomatically or with mild manifestations, sometimes the disease has severe manifestations, such as viral pneumonia that can evolve to respiratory failure and death in a short period of time. The natural evolution of infection by this virus includes an initial stage of respiratory epithelium infection and viral replication that can be followed by a second stage of immunopathology driven by a hyperinflammatory response that has systemic manifestations. The syndrome shares overlapping features with virus-induced hemophagocytic lymphohistiocytosis, including evidence of macrophage activation with a cytokine storm, and impairment of T lymphocyte and NK cell function. Understanding the pulmonary and extrapulmonary immunopathology of COVID-19 will enable the identification of biomarkers in an attempt to classify the disease as mild, moderate, severe, and critical, as well as for the development of new therapeutic strategies aimed at reducing generalized and pulmonary hyperinflammation in severe COVID-19.
Funding
The Laboratory of Immunobiology and Genetics of the National Institute of Respiratory Diseases Ismael Cosío Villegas received funding from the E022 fund for research from the Ministry of Health.
Conflicts of interest
The authors declare no conflicts of interest.
Ethical considerations
Protection of people and animals. The authors declare that no experiments were conducted on humans or animals for this research.
Confidentiality, informed consent, and ethical approval. The study does not involve personal data from patients nor require ethical approval. SAGER guidelines do not apply.
Declaration on the use of artificial intelligence. The authors declare that they did not use any type of generative artificial intelligence for writing this manuscript.
