Introduction
Lymphoid interstitial pneumonia (LIP) is a rare lymphoproliferative disorder of the pulmonary interstitial tissue classified as a rare disease according to the American Thoracic Society and the European Respiratory Society. Its diagnostic approach requires the use of multiple tools, including imaging studies (X-rays, computed tomography, etc.) and histopathological studies. This disease is characterized by the finding of a lymphocytic infiltrate that creates widening of the interlobular and alveolar septa1, commonly related to autoimmune disorders.
The case presented describes a 40-year-old patient, without relevant risk factors, who developed recurrent pneumothoraces as an atypical form of disease presentation, which underscores the significance of their clinical presentation. During the course, conventional treatment was given for the pneumothorax, as well as for the main suspected differential diagnoses, which were lymphangioleiomyomatosis and bullous disease2. Subsequently, treatment was redirected to the final diagnosis (LIP).
Case report
A 40-year-old woman with no significant past medical history presented in September 2023 with sudden onset of chest pain while sleeping, scoring 8/10 on the visual analog scale. The pain was described as sharp and radiating anteroposteriorly across the chest, with no clear trigger. It worsened with strenuous exertion and breathing. She denied cough, dyspnea, palpitations, syncope, sweating, nausea, and fever.
She was evaluated by a private physician, who prescribed symptomatic treatment, without apparent improvement. She subsequently went to a healthcare institution, where she was diagnosed with a 100% right pneumothorax through imaging studies. Intervention with a low-caliber pleural catheter3 was performed, leading to resolution of the condition without further diagnostic evaluation.
In January 2024, she presented a new episode of right pneumothorax, so she went to a private unit where a low-caliber pleural catheter was placed and chemical pleurodesis was performed without complications, with resolution of the condition. However, 3 months later, she presented a third right pneumothorax, which was the reason she came to our unit. She was again managed with a low-caliber pleural catheter, achieving resolution within 48 hours (Fig. 1A). The patient requested voluntary discharge after resolution of the condition; however, 13 days later she presented a new right pneumothorax (Fig. 1B). Given the recurrence, a low-caliber pleural catheter was placed to resolve the pneumothorax, and the patient was hospitalized with the aim of performing a diagnostic work-up due to the suspicion of probable lymphangioleiomyomatosis. Empirical treatment was initiated with sirolimus at a dose of 1 mg every 24 hours orally and budesonide/formoterol 160/4.5 µg inhaler (1 inhalation every 12 hours). Four days after admission, she developed a new right pneumothorax. A simple pulmonary computed tomography was requested, which revealed the presence of approximately 15 cystic formations at different levels (Figs. 1C–E). Laboratory studies did not show relevant alterations to the case.
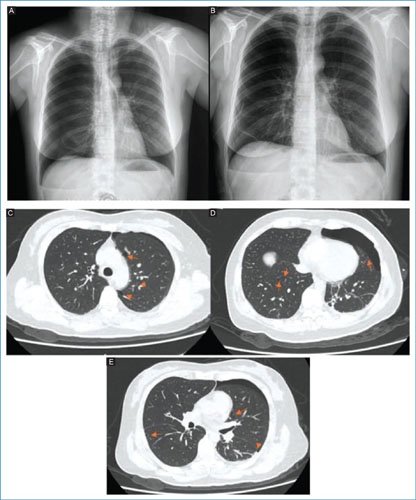
Figure 1. A: right spontaneous pneumothorax is observed. B: right pulmonary expansion with Heimlich valve catheter is highlighted. C: the presence of multiple pulmonary bullae is noted. D: presence of left pneumothorax with multiple bullae in both lungs. E: presence of bullae at the bilateral basal pulmonary level.
A sample was taken from a cystic lesion for histopathological study. Subsequently, pleurodesis and placement of a 28 Fr pleural tube with suction were performed, with good clinical progress. The tube was removed at 48 hours. The patient remained under observation for 5 days after removal, without experiencing a new episode of spontaneous pneumothorax. The histopathological study reported lesions suggestive of LIP (Fig. 2). Treatment with sirolimus was discontinued, and management with systemic and inhaled steroids was initiated. To date, the patient has shown a favorable progress without new episodes of spontaneous pneumothorax, and continues to be evaluated for a probable associated autoimmune pathology.
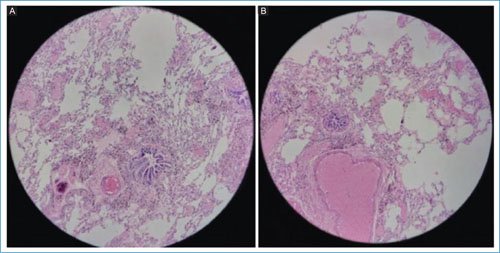
Figure 2. Pulmonary biopsy in a patient with a diagnosis of lymphoid interstitial pneumonia. A: thickening of the alveolar interstitium is observed due to a diffuse lymphoid infiltrate with peribronchiolar and perivascular disposition, associated with the presence of reactive germinal centers. Areas of partial occupation of the alveolar spaces by inflammatory cells are also identified. B: more prominent lymphoplasmacytic infiltrate in the interstitium with the formation of well-organized lymphoid follicles. The relative preservation of pulmonary architecture and the absence of necrosis or granulomas stand out, characteristic findings of LIP.
Discussion
A clinical case is presented in which a rare pathology in the medical field is diagnosed, about which much remains to be discovered regarding its etiology and treatment, which remain uncertain. The patient exhibited a rare clinical presentation within pulmonary pathologies due to LIP by presenting a total of five spontaneous pneumothoraces in less than 6 months, making it a case of importance for study. The patient showed no laboratory alterations nor had a relevant history that would help with probable diagnostic suspicions, which created a diagnostic challenge. Due to the recurrent pneumothoraces, the patient’s age, and without other relevant information, a lymphoproliferative disease, such as lymphangioleiomyomatosis (a rare disease), was initially suspected, which is characterized by recurrent pneumothoraces as a clinical presentation4, and therefore, treatment based on sirolimus was initiated. Upon correcting the diagnosis by the histopathological method, treatment was corrected. Currently, there is no specific treatment for LIP, so it is a difficult pathology to control and to have continuous follow-up5.
Since LIP is a rare disease, its prevalence and incidence are not known with certainty6. According to the literature, it represents less than 1% of all pulmonary diseases, making the presented case important. It is known to have a tendency for presentation around 40 years of age and a predilection for women (2.75:1)7. Current data come mainly from retrospective studies and isolated case reports, which limit our understanding of its true prevalence and incidence in different populations. It is reported that the common symptomatology of LIP is progressive dyspnea with dry cough, occasionally accompanied by pleuritic-type pain and systemic symptoms such as fever, weight loss, fatigue, and night sweats. It is sometimes asymptomatic, and the diagnosis is established incidentally7. Little reference is made to the appearance of pneumothorax, with our case being a very atypical form of presentation compared to the current literature.
The diagnostic approach requires imaging studies such as pulmonary computed tomography, which shows characteristics of cystic lesions, ground-glass opacities, etc. Among the differential diagnoses, Langerhans cell histiocytosis shows irregular cysts with thick walls and predominance in the upper lobes, with centrilobular nodules. This apical distribution and the presence of nodules are distinctive features compared to LIP. Pneumocystosis can generate cysts associated with ground-glass opacities, which are frequent in immunosuppressed patients. However, in LIP, the inflammatory infiltrates are more persistent and usually predominate in the absence of immunosuppression. Birt-Hogg-Dubé syndrome presents rounded or oval cysts with thin walls, located mainly in the lung bases and subpleural areas; unlike LIP, these cysts are usually isolated and are not accompanied by inflammatory infiltrates. Finally, in autoimmune diseases such as Sjögren’s syndrome and scleroderma, pulmonary cysts are often associated with subpleural fibrosis and a honeycomb pattern8. In a histological study, they are characterized by a diffuse lymphocytic infiltrate in the alveolar walls, composed of mature lymphocytes, plasma cells, and occasionally eosinophils. A distinctive finding is lymphoid follicles with germinal centers, around the airways and blood vessels. There is reactive hyperplasia of type II pneumocytes and pulmonary cysts due to alveolar damage. Malignancy should not be observed9.
Known causes of LIP are autoimmune diseases (systemic lupus erythematosus, Sjögren’s syndrome, etc.) or viral infections (human immunodeficiency virus, Epstein-Barr virus, etc.). However, between 10% and 25% of LIP cases do not have an identifiable cause and are classified as idiopathic; this percentage varies according to populations and available diagnostic tools10.
Recommended treatments include corticosteroids and treatment of the underlying cause. Currently, the patient continues in the research protocol without presenting new episodes of spontaneous pneumothorax. Continuous surveillance is recommended, as few cases of disease progression to neoplasms such as lymphomas have been reported9.
Conclusions
The presented clinical case demonstrates an extremely rare manifestation of LIP, characterized by multiple episodes of spontaneous pneumothorax in a patient without risk factors or relevant history. The complexity of the initial diagnosis and the atypicality of the clinical presentation underscore the need to integrate histopathological studies as an essential part of the approach to cases with uncommon characteristics.
The final diagnosis allowed for adjustment of clinical management, highlighting the fundamental role of corticosteroids in treatment and long-term follow-up to prevent complications. This case not only reinforces the importance of maintaining a comprehensive and systematic approach in the evaluation of pulmonary diseases but also opens the door to future research, especially regarding unusual manifestations such as recurrent pneumothorax. Multidisciplinary collaboration and the development of broader registries on this disease will be crucial for improving the understanding and treatment of LIP.
Funding
None.
Conflicts of interest
The authors declare that they have no conflicts of interest.
Ethical considerations
Protection of persons and animals. The authors declare that no experiments were conducted on humans or animals for this research.
Confidentiality, informed consent, and ethical approval. The authors have followed their institution’s confidentiality protocols, have obtained informed consent from the patients, and have the approval of the Ethics Committee. The recommendations of the SAGER guidelines have been followed, according to the nature of the study.
Declaration on the use of artificial intelligence. The authors declare that they did not use any type of generative artificial intelligence for the writing of this manuscript.
