Introducción
La COVID-19, asociada al SARS-CoV-2, ha causado millones de muertes en todo el mundo. Desafortu-nadamente, la pandemia que surgió en 2019 en la provincia China de Wuhan se ha extendido por varios años y han aparecido nuevas variantes del virus, como ómicron y algunas variantes de esta. Al principio de la pandemia, en febrero del año 2020, en el Instituto Nacional de Enfermedades Respiratorias Ismael Cosío Villegas (INER) se diagnosticó uno de los primeros casos confirmados para este virus en México, aunque muy probablemente ya existían otros casos que pudieron coexistir con este caso índice en el sur de la Ciudad de México. Un acierto de las autoridades y las instituciones, como el INER y otros institutos nacionales de salud de México, fue la anticipación y la preparación en respuesta a los informes de este brote que rápidamente se extendió por China y otros países, y una alerta que fue emitida en las últimas semanas del año 2019 por las autoridades de la Secretaría de Salud de México. Lo anterior permitió establecer estrategias operativas de diagnóstico y manejo de casos de enfermedad respiratoria por coronavirus y restablecer los sistemas de referencia y confirmación de muestras mediante técnicas de biología molecular. En ese momento, el INER, por mandato de las autoridades, se reconvirtió a un hospital específicamente dedicado al diagnóstico y el manejo de casos graves de COVID-19. Sin embargo, esto no ocurrió de forma paralela en otros niveles de atención. La falta de diagnóstico temprano y de confinamiento de nuevos casos y sus contactos, en ese momento fueron factores asociados a unas mayores morbilidad y mortalidad. A la fecha sigue siendo un reto de salud pública detener las cadenas de transmisión del SARS-CoV-2. La falta de acceso a antivirales específicos y a vacunas también ha contribuido de manera significativa al desarrollo de formas graves y desenlaces clínicos pobres en los pacientes con COVID-19.
La gran cantidad de investigaciones realizadas y de literatura sobre las características del SARS-CoV-2, su origen, sus mecanismos de infección, replicación y tropismo celular, así como sobre las características del huésped y los factores de riesgo de infección y de desarrollo de formas graves de la enfermedad, ha permitido el desarrollo de una serie de estrategias profilácticas, terapéuticas y de control de la infección con una rapidez jamás vista en la historia de la medicina aplicada. A pesar de esto, la comprensión de los factores del virus y del huésped que determinan el comportamiento clínico de la enfermedad aún es incompleta. Se han identificado, mediante sofisticados análisis de expresión del genoma, firmas moleculares asociadas con la enfermedad grave que a la fecha se utilizan como biomarcadores de pronóstico para orientar las decisiones terapéuticas1–8.
Se han identificado los mecanismos de la respuesta inmunitaria, tanto de la respuesta innata de células como los macrófagos como de la respuesta adaptativa de linfocitos T y B que determinan respuestas inflamatorias poco reguladas que pueden contribuir al daño a los tejidos. Un ejemplo de ello son las evidencias histopatológicas de hemofagocitosis en la médula ósea y órganos reticuloendoteliales, y el síndrome de activación de macrófagos observado en la linfohistiocitosis hemofagocítica inducida por este virus, lo cual apunta a que el sistema inmunitario innato del huésped tiene un papel crucial en la inmunopatología de la COVID-19. De hecho, diversos estudios han destacado la eficacia de algunos agentes inmunomoduladores que permiten atenuar la respuesta inflamatoria al SARS-CoV-2 y evitar la lesión pulmonar.
Es importante destacar que se observó una incidencia cada vez mayor de secuelas en pacientes convalecientes de COVID-19, incluida la fibrosis pulmonar, especialmente entre los que se recuperaron de una enfermedad grave. Estas complicaciones podrían afectar de forma permanente la función respiratoria de los pacientes, impactando negativamente en su calidad de vida. Por lo anterior, es prioritario estudiar los procesos patogénicos subyacentes a la inflamación excesiva, la lesión tisular y los mecanismos de remodelación de los tejidos, incluyendo la matriz extracelular, después de la infección por SARS-CoV-2.
En esta revisión presentamos una perspectiva general de los mecanismos de infección del SARS-CoV-2 y de los procesos inmunitarios de protección y daño asociados a las diferentes formas clínicas de la COVID-19.
Biología del SARS-CoV-2
El SARS-CoV-2 es un miembro del grupo de coronavirus humanos (HCoV), constituido por HCoV-229E, HCoV-NL63, HCoV-HKU1, HCoV-OC43, SARS-CoV-1 y MERS-CoV1. Estos patógenos son coronavirus de RNA de cadena única que pertenecen a la familia Coronaviridae. Algunos de ellos han causado una variedad de enfermedades respiratorias de diferente gravedad en el pasado; por ejemplo, el SARS-CoV-1 infectó a más de 8000 personas en Asia en 20032, y el MERS-CoV (coronavirus relacionado con el síndrome respiratorio de Medio Oriente) originado en Arabia Saudita en 2012 tuvo unas altas tasas de mortalidad3,4.
Mediante estudios de comparación de la secuencia de los genomas virales, los HCoV pueden agruparse en cuatro géneros: alfa, beta, gamma y delta. El nuevo SARS-CoV-2 es un betacoronavirus genéticamente relacionado con un coronavirus del murciélago llamado BatCoV RaTG13, así como con el SARS-CoV-15,6. Además, el SARS-CoV-2 comparte identidad genética con algún coronavirus aislado de pangolines7,8. Por lo tanto, se cree que la COVID-19 es una enfermedad zoonótica originada en el murciélago y con el pangolín como posible huésped intermediario. El genoma del SARS-CoV-2 consiste en una cadena de RNA de 29,903 pares de bases que codifica para una replicasa-transcriptasa y para las proteínas estructurales Spike (S), de envoltura (E), de membrana (M) y nucleocápside (N)5.
Mecanismo de infección por el SARS-CoV-2
El paso inicial en el proceso de infección por el SARS-CoV-2 es el reconocimiento de su receptor en la membrana de las células del huésped. Este proceso es mediado por la proteína S, la cual reconoce la enzima convertidora de la angiotensina 2 humana (ACE2, angiotensin converting enzyme 2), el mismo receptor para la proteína S del SARS-CoV-19–11. Esta proteína tiene dos dominios funcionales: el dominio S1 contiene el sitio de unión al receptor (RBD, receptor binding domain) que se fija a ACE2, mientras que el domino S2 realiza la fusión de la membrana viral con la membrana de la célula blanco11. Por lo tanto, la distribución del receptor ACE2 en diferentes órganos y tejidos es crucial para determinar la infectividad y el tropismo del virus.
Un aspecto muy importante del proceso de infección es la activación de la proteína S. Dicho proceso es mediado por diferentes proteasas del huésped que rompen la proteína S en sus sitios S1/S2 y S’2. Este procesamiento proteico permite una completa actividad funcional al dominio S2 de la proteína S para que pueda fusionar las membranas viral y celular. Para dicho propósito, tal como lo hace el SARS-CoV-1, el SARS-CoV-2 emplea la proteasa 2 de serina transmembrana (TMPRSS2, transmembrane protease serine 2)9,12,13. De forma interesante, las proteasas TMPRSS4 y catepsina L14,15, así como el receptor humano CD14716, también promueven la infección por el SARS-CoV-2. Por lo tanto, el patrón de expresión tisular de estos elementos podría ser decisivo del tropismo del virus, e incluso algunos fármacos que inhiben la actividad de dichas proteasas o del receptor CD147 se han propuesto como agentes terapéuticos para prevenir y tratar la COVID-199,15,16.
Otro factor involucrado en el proceso de infección por SARS-CoV-2 es la cinasa de fosfatidilinositol-3-fosfato (PIKfyve, phosphoinositide kinase, FYVE-type)15. Esta enzima regula la producción de fosfatidilinositol-3,5-bifosfato, un fosfolípido que participa en el proceso de maduración de los endosomas. De hecho, el apilimod, un potente inhibidor de PIKfyve, reduce la infectividad del SARS-CoV-2 y podría ser un candidato para propósitos terapéuticos15.
Una vez que el virus logra ingresar a las células comienza la replicación viral con la traducción del gen de la replicasa-polimerasa y el ensamble del complejo de replicación-transcripción. Este complejo transcribe las regiones genómicas del virus que codifican para las proteínas estructurales. De esta forma, nuevos viriones son producidos en el retículo endoplásmico y el aparato de Golgi para salir finalmente de la célula1. Una característica particular del SARS-CoV-2 es que posee una secuencia polibásica de escisión por furina en el sitio S1/S2, ausente en otros coronavirus relacionados6,11. Esta secuencia es procesada en el aparato de Golgi durante la biosíntesis de la proteína S de los viriones nuevos11. Por lo tanto, los nuevos viriones de SARS-CoV-2 poseen una proteína S ya activada y lista para unirse al receptor ACE2, sin requerir la actividad de otras proteasas del huésped. Dado que virtualmente todas las células humanas expresan furina, la inserción de una secuencia de escisión por furina podría expandir la capacidad de transmisión y tropismo del SARS-CoV-2.
Respuesta inmunitaria contra el SARS-CoV-2
Se sabe que una vez que el SARS-CoV-2 logra su primera fase de replicación en las vías respiratorias altas se disemina de forma temprana hacia el tracto respiratorio bajo, en donde se desencadena una respuesta inmunitaria innata pronunciada que lleva, en los casos sintomáticos, al inicio de las manifestaciones clínicas. Entre los signos y síntomas más frecuentes observados en personas con COVID-19 se encuentran fiebre, tos seca, fatiga, cefalea, diarrea, disnea, anosmia y pérdida del gusto17,18. Sin embargo, la enfermedad tiene un espectro clínico heterogéneo que incluye casos asintomáticos, enfermos con manifestaciones leves y pacientes con síntomas moderados a graves que desarrollan insuficiencia respiratoria aguda, disfunción multiorgánica y riesgo de fatalidad19,20. Afortunadamente, un 85% de las personas infectadas con SARS-CoV-2 no presentan manifestaciones clínicas o sufren una enfermedad leve, mientras que solo un 5-30% de los casos pueden presentar una forma crítica21–23. Lo anterior quiere decir que, en la vasta mayoría de los casos de COVID-19, el sistema inmunitario es eficaz en controlar la infección y eliminar el virus. No obstante, como ya fue mencionado, los componentes inmunitarios que participan en las respuestas protectoras contra el SARS-CoV-2 aún se encuentran en estudio.
Clásicamente, se cree que la defensa contra una infección viral involucra la activación de mecanismos como la producción de interferones tipo I, los cuales limitan la replicación del patógeno dentro de las células infectadas y previenen su diseminación a células sanas. Asimismo, para neutralizar a las partículas virales se requiere una respuesta humoral robusta con producción de anticuerpos específicos de antígeno de alta afinidad. Finalmente, la inmunidad celular desempeña un papel clave en la identificación y la lisis de células infectadas para eliminar los reservorios intracelulares del patógeno. Todos estos mecanismos podrían ser importantes para la protección contra la COVID-19, por lo que a continuación se resume la evidencia actual sobre el rol de diferentes factores inmunitarios durante la infección por SARS-CoV-2.
Respuesta inmunitaria innata
El sistema inmunitario innato provee protección inespecífica contra una amplia variedad de microorganismos. Entre sus componentes principales se encuentran las barreras epiteliales y mucosas, además de factores humorales y celulares que en conjunto pueden reconocer a los patógenos e iniciar una respuesta inflamatoria que, en la mayoría de los casos, es suficiente para eliminar la infección. Las barreras mucosas, como el epitelio respiratorio, cuentan con una serie de mecanismos que impiden la adhesión de los patógenos a la superficie de las células epiteliales y el inicio de la infección. La importancia de la mucosa respiratoria radica en que cualquier alteración de su integridad, como en el caso de personas con enfermedades bronquiales y pulmonares crónicas, confiere mayor riesgo de contraer COVID-19 y presentar una forma grave de la enfermedad.
Entre los mecanismos de barrera que protegen al huésped se encuentra la producción de moco en las vías respiratorias, que atrapa y permite la eliminación de virus y bacterias, así como la producción de surfactante en el epitelio alveolar, el cual contiene algunos factores proteicos de defensa que permiten neutralizar a diferentes microorganismos. Tal es el caso de la proteína del surfactante D (SP-D, surfactant protein D), la cual puede unirse a la proteína S del SARS-CoV-2 y neutralizarla, inhibiendo la entrada del virus a las células epiteliales24. No obstante, algunos estudios sugieren que, en casos graves de la enfermedad, el virus es capaz de contrarrestar los efectos del surfactante mediante la supresión de la producción de SP-D25,26. Otros elementos moleculares producidos por el epitelio respiratorio y que son suprimidos durante la infección incluyen algunas citocinas con propiedades antimicrobianas, como la quimiocina CXCL1727.
Una vez que el SARS-CoV-2 logra superar los mecanismos de barrera, el siguiente paso en la defensa contra el virus incluye el reconocimiento de patrones moleculares asociados a patógenos, como la cadena única de RNA viral o las proteínas de envoltura, por parte de receptores de reconocimiento de patrones de superficie o intracelulares que se expresan en células epiteliales, macrófagos alveolares y otras células del sistema fagocítico-mononuclear. Actualmente, aún no se conocen todos los receptores inmunitarios innatos que reconocen la infección viral e inician las respuestas inmunitarias contra el SARS-CoV-2. Dado que este virus está genéticamente relacionado con el SARS-CoV-1, se presume que ambos comparten mecanismos de infección. En este sentido, el SARS-CoV-1 es reconocido por los receptores tipo Toll (TLR, Toll like receptors) TLR3 y TLR4, que inducen una reacción inmunitaria a través de las vías MyD88 y TRIF28,29. Además, el SARS-CoV-1 desencadena la producción de interleucina (IL) 1β a través de la activación del inflamasoma30, con el objetivo de desencadenar una respuesta inflamatoria contra el virus. A este respecto, las investigaciones recientes indican que el TLR2 es otro de los sensores innatos capaces de reconocer al virus mediante su unión a la proteína de envoltura. Una vez que ocurre dicho reconocimiento, el TLR2 induce una reacción inflamatoria a través de la vía de señalización dependiente de MyD8831. La activación del inflamasoma también es posible que ocurra durante la infección por SARS-CoV-2, ya que se han observado altos niveles de IL-1β en pacientes con COVID-19, como se describe más adelante32. Los estudios han demostrado que la infección por SARS-CoV-2 induce la activación de inflamasomas NLRP3 y la liberación de IL-1β en cultivos de monocitos y en células mononucleares de sangre periférica de pacientes con COVID-19; no obstante, una activación exagerada de los inflamasomas también puede llevar al desarrollo de formas graves de la enfermedad33,34.
Otro mecanismo innato crucial en la defensa contra las infecciones virales es la producción de interferones (IFN) de tipo I (IFN-α e IFN-β, principalmente), los cuales se unen a complejos de receptores en superficies de membrana conocidos, como el receptor IFN-α/β(IFNAR), que consiste en cadenas de IFNAR1 e IFNAR2. Una vez unidos a su receptor, los IFN tipo I pueden interferir en la replicación de los virus en las células hospedadoras, activando diferentes vías de señalización en las que participan proteínas antivirales como la PKR. Los estudios in vitro indican que la actividad de los IFN tipo I es eficaz para inhibir la replicación del SARS-CoV-2 en células humanas35. En modelos de COVID-19 en primates se ha observado que la respuesta mediada por IFN es ampliamente inducida en los pulmones y resulta crucial en la defensa contra la infección36. Sin embargo, es posible que la supresión de IFN tipo I sea una estrategia del SARS-CoV-2 para evadir la respuesta inmunitaria innata antiviral, ya que algunos estudios han mostrado muy baja producción de IFN-α e IFN-β en el plasma de pacientes mexicanos y franceses con formas graves de COVID-1937,38.
Finalmente, algunas células del sistema inmunitario innato podrían desempeñar una función importante en la defensa del huésped. Entre estas células se encuentran los macrófagos alveolares, los cuales se presume que son las primeras células del sistema inmunitario en entrar en contacto con el SARS-CoV-2. Aunque estas células pueden reconocer al virus, su respuesta es manipulada por el patógeno39,40. Otras células cruciales en la defensa contra las infecciones virales son las células NK (natural killer), pero su función en la COVID-19 aún no es por completo conocida. En general, la mayoría de los estudios en los que se ha caracterizado el perfil inmunológico de pacientes con COVID-19 muestran que las células NK se encuentran disminuidas en la sangre de aquellos con enfermedad grave41,42. Se cree que algunas deficiencias en la expresión de receptores de células NK, como el NKG2C, podrían asociarse a mayor riesgo de sucumbir a la infección43. Lo anterior sugiere un posible papel protector de las células NK en la respuesta inmunitaria contra el SARS-CoV-2.
Respuesta inmunitaria adaptativa
El sistema inmunitario adaptativo provee una segunda línea de defensa contra patógenos orquestada por linfocitos que poseen receptores específicos de antígenos, capaces de someterse a recombinación genética. Por lo tanto, la inmunidad adaptativa contra los virus es capaz de controlar la infección de manera eficaz en la mayoría de los casos, a través de mecanismos dirigidos específicamente al patógeno agresor. Las células de la inmunidad adaptativa pueden promover respuestas protectoras contra patógenos mediante la producción de citocinas proinflamatorias y antivirales por parte de los linfocitos T cooperadores CD4+, o bien a través de la destrucción de células infectadas mediada por linfocitos T citotóxicos CD8+. Se cree que estas células podrían desempeñar un papel fundamental en la defensa contra el SARS-CoV-2, pues se ha observado que, en otras infecciones por coronavirus, los linfocitos T específicos de antígeno son cruciales para conferir protección. De hecho, en personas que fueron afectadas por el SARS-CoV-1 en 2003 y sobrevivieron a la enfermedad, se ha comprobado que diferentes clonas de linfocitos T específicos de memoria sobreviven y son capaces de responder al virus hasta 17 años después de la primoinfección44. Algunos estudios sugieren que los individuos infectados por SARS-CoV-2 desarrollan linfocitos T CD4+ y CD8+ de memoria, los cuales persisten en la circulación hasta por 8 meses45. Estas respuestas de memoria también incluyen linfocitos T foliculares, los cuales son cruciales para apoyar a los linfocitos B en la producción de anticuerpos protectores. Pese a lo anterior, los datos actuales sobre el papel de la inmunidad celular en la patogénesis de la COVID-19 aún son poco claros y resultan controversiales. Por ejemplo, algunos investigadores han encontrado que los pacientes con formas graves de la enfermedad tienen números reducidos de linfocitos T en la circulación, pero los linfocitos T específicos de antígeno de dichos individuos desarrollan respuestas más potentes de producción de citocinas46.
La respuesta humoral también es importante para controlar las infecciones virales en los humanos. En este sentido, las células B de individuos infectados por SARS-CoV-2 experimentan un cambio de clase de anticuerpos de inmunoglobulina (Ig) M a IgG e IgA de mayor afinidad. Así, los sueros de pacientes convalecientes de COVID-19 contienen anticuerpos IgG, IgM e IgA antiproteína S y anticuerpos IgG antinucleocápside. No obstante, se han encontrado títulos elevados de anticuerpos IgG e IgM en pacientes con enfermedad grave, lo que cuestiona la capacidad protectora de las respuestas de anticuerpos en la COVID-1947. Es importante abordar la capacidad neutralizante de los anticuerpos inducidos por la infección natural y la vacunación, especialmente para mejorar la capacidad protectora de la respuesta humoral provocada por las vacunas. Un estudio de Dan et al.45 revela que incluso cuando hay distintas cinéticas de anticuerpos neutralizantes contra diferentes componentes del SARS-CoV-2, las células B de memoria específicas de antígeno permanecen detectables durante más de 8 meses después de la aparición de los síntomas en algunos pacientes con COVID-19. Ellebedy et al. han observado diferentes clonas de células plasmáticas de vida larga y linfocitos B de memoria en la médula ósea y la sangre de personas que se recuperaron de COVID-19 hasta 11 meses después de la infección. Estas células plasmáticas y los linfocitos B de memoria pueden responder a encuentros secundarios con el SARS-CoV-2 y producir anticuerpos neutralizantes48. Por último, la importancia de las respuestas de memoria inmunitaria adaptativa mediadas por linfocitos B para la defensa contra la COVID-19 también es señalada en estudios que han demostrado que la reacción de centros germinales en los ganglios linfáticos se encuentra abolida o alterada en personas gravemente enfermas tras la infección por SARS-CoV-249.
Los mecanismos de defensa contra el SARS-CoV-2 se resumen en la figura 1.
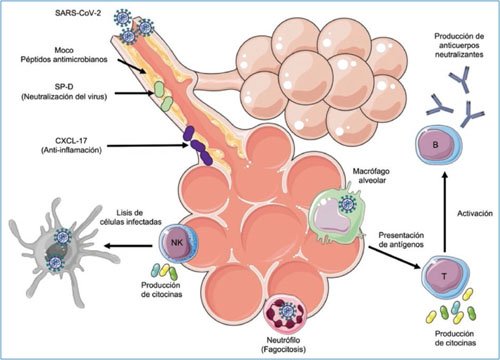
Figura 1. Mecanismos de defensa inmunitaria contra el SARS-CoV-2. NK: célula asesina natural; SP-D, proteína del surfactante D.
Inmunopatología de las formas graves de COVID-19
Tormenta de citocinas e inmunosupresión
Las personas que progresan a COVID-19 grave presentan neumonía dentro de los 10-20 días posteriores al inicio de los síntomas, que se asocia con una reducción de la saturación de oxígeno, síndrome de insuficiencia respiratoria aguda y daño pulmonar prominente con opacidades en vidrio deslustrado. El síndrome de insuficiencia respiratoria aguda se caracteriza por una mayor permeabilidad pulmonar, hipoxemia grave y edema pulmonar no cardiogénico. Estas condiciones alteran la barrera alveolocapilar y son consecuencia de un proceso de hiperinflamación sistémica50–52.
Para guiar las intervenciones inmunoterapéuticas en pacientes con condiciones críticas es crucial una mejor comprensión de los factores del huésped implicados en la inmunidad protectora, frente a la patogénica, contra el SARS-CoV-2. Desafortunadamente, lo que hoy comprendemos sobre la inmunopatología de la COVID-19 grave es una paradoja: la respuesta adaptativa es hiperactiva, pero incapaz de controlar al virus. De hecho, los pacientes con COVID-19 presentan un perfil de citocinas proinflamatorias (IL-1β, IL-2, IL-6, IL-7, IL-8, IL-9, FGF, G-CSF, GM-CSF, IFN-g, CXCL10, CCL2, CCL3, CCL4, PDGF, TNF-α y VEGF) y reguladoras (IL-10 y TGF-β)21,53–55. Este fenómeno se ha denominado «tormenta de citocinas» y media el daño tisular en pacientes con COVID-19 que progresan a enfermedad grave55–58. De estos factores, IL-1β, IL-6, CXCL10 y TNF-α son las citocinas con mayor capacidad de generar daño tisular en diferentes órganos, incluyendo el cerebro, debido a sus propiedades inflamatorias. Por ejemplo, IL-1β e IL-6 se han implicado en la neurotoxicidad asociada con la terapia con células T de receptor de antígeno quimérico en pacientes con neoplasias hematológicas59,60. Estas citocinas poseen efectos perjudiciales sobre la función endotelial en varios nichos vasculares, que pueden estar implicados en la fisiopatología de las complicaciones trombóticas y neurológicas de la COVID-19. En este sentido, es importante mencionar que otros órganos, además de los pulmones, también pueden verse afectados por el SARS-CoV-2, como el cerebro, el corazón, los intestinos, los riñones y el hígado, entre otros.
Esta reacción inflamatoria generalizada también induce la producción de varios reactantes de fase aguda, como la proteína C reactiva y la ferritina en el hígado, que aumentan aún más la liberación de mediadores inflamatorios y enriquecen la liberación de citocinas en pacientes con COVID-19 grave61,62. De hecho, los niveles elevados de ferritina predicen el riesgo de muerte en estos pacientes62. Además, la hiperinflamación observada en pacientes con COVID-19 en estado crítico puede asociarse con el desarrollo de eventos trombóticos. En este sentido, los parámetros de coagulación anormales, como unos niveles más altos de dímero D y unos tiempos de protrombina y de tromboplastina parcial activada más prolongados, se han asociado con un pronóstico desfavorable. Estos parámetros de coagulación anormales se producen poco después de la hospitalización, y en algunos pacientes, las concentraciones de fibrinógeno y la actividad de la antitrombina disminuyen con el tiempo63–65.
Curiosamente, a diferencia de otros síndromes de tormenta de citocinas, la activación inmunitaria polifuncional de la COVID-19 se acompaña de linfocitopenia y una fuerte infiltración de células inmunitarias, primordialmente mononucleares (linfocitos y macrófagos), en el intersticio del pulmón66–68. En los pacientes con infección grave por SARS-CoV-2, una amplia gama de subtipos de células inmunitarias se encuentra disminuida en la circulación. Estas células incluyen monocitos, células dendríticas, células T CD4 + y CD8 +, y células NK41. Además, los pocos linfocitos adaptativos que quedan en la sangre expresan marcadores de agotamiento funcional69. Estos datos sugieren que la COVID-19 grave produce un estado de inmunosupresión similar a la inducida por la sepsis70; también es posible que el reclutamiento robusto de células inmunitarias funcionales a los sitios de infección por el SARS-CoV-2 pueda explicar la leucocitopenia observada durante la COVID-19. Un estudio reciente de Remy et al.71 ha demostrado que la inmunosupresión observada en la COVID-19 es incluso más profunda que la de pacientes críticamente enfermos con sepsis de otras causas. Estos investigadores observaron que la producción de IFN-γ por las células T de sangre periférica de pacientes con COVID-19 se vio afectada en comparación con las células T de individuos sanos y pacientes con sepsis después de la estimulación con anticuerpos anti-CD3/anti-CD28; se encontró una producción reducida de TNF-α por los monocitos estimulados de pacientes con COVID-19. Estos hallazgos llevaron a los investigadores a proponer que el mecanismo inmunitario primario subyacente a la morbilidad y la mortalidad de la COVID-19 es la inmunosupresión en lugar de la hiperinflamación.
El perfil inmunitario en la COVID-19 grave tiene características únicas que la diferencian de otras infecciones respiratorias, como la influenza A (H1N1) pandémica. Entre los factores inmunitarios que solo se encuentran en pacientes críticos con COVID-19, y no en pacientes con influenza, se encuentran IFN-γ, IL-4, IL-5, IL-6, IL-10, IL-12, IL-13, IL-1β, CCL11, VEGF, TWEAK, TSLP, MMP-1 y MMP-338. Estas moléculas podrían desempeñar un papel específico en la COVID-19 y son objetivos potenciales para reducir su morbilidad y mortalidad. Es de destacar que los niveles más altos de citocinas Th2, particularmente IL-4 e IL-5, podrían inhibir las respuestas antivirales protectoras Th1 en los pacientes con COVID-19. Por lo tanto, la falta de equilibrio inmunitario del tipo de respuesta efectora es otro determinante crucial del colapso de la inmunidad protectora del huésped contra el SARS-CoV-2. Esta respuesta sesgada Th2 puede generar infiltrados intersticiales de células Th2, neutrófilos, eosinófilos y células linfoides innatas de tipo 2, que median la inflamación pulmonar y el daño tisular. Los pacientes con COVID-19 críticamente enfermos suelen mostrar infiltrados pulmonares intersticiales, algunos de los cuales se asemejan a varias formas de enfermedad pulmonar intersticial progresiva, como la neumonía organizada criptogénica y la neumonía intersticial no específica21,72–74. Estos efectos nocivos de las respuestas Th2 también podrían explicar las anomalías en la función pulmonar y la progresión a fibrosis pulmonar observada en más del 45% de los pacientes con COVID-19 dados de alta de los hospitales75, particularmente sujetos ancianos. Por lo tanto, sería de gran interés caracterizar el perfil de citocinas de los pacientes con COVID-19 que posteriormente desarrollan cualquier forma de enfermedad pulmonar intersticial, ya que se beneficiarían de terapias específicas antifibróticas.
Infiltración de neutrófilos y daño pulmonar
Una variedad de mediadores inflamatorios liberados de forma exagerada durante la infección grave por SARS-CoV-2 tienen como función mediar el reclutamiento de células mieloides hacia los sitios de inflamación. Unas de las células que infiltran en forma extensa los pulmones de pacientes con COVID-19 grave son los neutrófilos, probablemente en dirección hacia un gradiente quimiotáctico generado por las altas concentraciones de IL-8, CXCL10, CCL2 y CCL3 liberadas a la circulación21,53–55. Los neutrófilos son los leucocitos más abundantes en la sangre y tiene una vida media corta. Debido a sus características móviles, pueden infiltrar fácilmente tejidos inflamados y contribuir a la respuesta de defensa contra diferentes microorganismos. Su función principal es mediar la fagocitosis de patógenos, así como la liberación de citocinas y sustancias proteolíticas contenidas en sus gránulos. Los neutrófilos pueden ejercer una función protectora en la defensa contra infecciones virales76; es bien sabido que los neutrófilos siempre están presentes en los pulmones de pacientes con diferentes infecciones respiratorias asociadas al desarrollo de síndrome de dificultad respiratoria aguda, especialmente debido a su capacidad de desgranulación y lisis, promoviendo el daño tisular.
Consistente con lo anterior, en modelos animales de macacos se ha observado que los neutrófilos infiltran el pulmón rápidamente durante la infección por SARS-CoV-236. Esto también se observa en pacientes con COVID-19, en quienes se ha encontrado que la neutrofilia y un índice neutrófilo:linfocito más alto es una característica distintiva de los casos graves y de aquellos que se presentan en pacientes ancianos, asociado a un pronóstico desfavorable36,77. En pacientes fallecidos por COVID-19 grave se ha observado una intensa infiltración de neutrófilos en los capilares pulmonares, extravasación hacia los espacios alveolares y mucositis neutrofílica en las vías respiratorias78. Dentro de los sitios de infección por SARS-CoV-2, la desgranulación de los neutrófilos resulta en la liberación de proteasas con capacidad de amplificar la respuesta inflamatoria. Tal es el caso de la catepsina G, la cual estimula una mayor producción de citocinas y quimiocinas, promoviendo el reclutamiento de monocitos, macrófagos y neutrófilos, así como el aumento de la permeabilidad endotelial y epitelial36,79. La catepsina G también activa algunas metaloproteinasas de matriz (MMP)80, dando inicio a la alteración de los mecanismos que regulan la composición de la matriz extracelular (MEC) y se asocian a la aparición de secuelas características del síndrome post-COVID-19, como se describe más adelante. Finalmente, los neutrófilos son fuente de un exceso de trampas extracelulares, las cuales exacerban aún más la tormenta de citocinas y perpetúan un círculo vicioso de inflamación y reclutamiento de neutrófilos hacia el pulmón78. Por lo tanto, está justificado que la búsqueda de nuevas estrategias para reducir la morbimortalidad de la COVID-19 se enfoque en la interrupción del reclutamiento de neutrófilos hacia el pulmón y la limitación del daño pulmonar inducido por el contenido de los gránulos de dichas células.
Mecanismos inmunitarios asociados al síndrome post-COVID-19
El síndrome post-COVID-19 o secuelas posagudas de la COVID-19 (PASC, post-acute sequelae of COVID-19) se define por la persistencia de la enfermedad durante más de 28 días después del inicio de los síntomas. Diversos estudios longitudinales sugieren que este proceso puede observarse entre el 30% y el 80% de los individuos que sufren infección por el SARS-CoV-2. En este sentido, una característica notable de la fase de convalecencia de la COVID-19 es que muchos pacientes que sufrieron daño pulmonar grave permanecen con disfunción pulmonar permanente. Muchos de los efectos deletéreos crónicos de la COVID-19 están relacionados con la fibrosis pulmonar posterior a la lesión. Los mecanismos subyacentes al desarrollo de estas complicaciones no han sido bien definidos hasta ahora. En pacientes con neumonía grave de otras etiologías, con frecuencia se ha observado daño epitelial y endotelial grave, acompañado de fibrosis extensa. Los pacientes que presentan mayores cambios fibróticos son aquellos que requirieron periodos prolongados de ventilación mecánica (~12 días) y desarrollaron insuficiencia orgánica sistémica más grave50,81. De forma interesante, la fibrosis pulmonar también es una secuela a lo largo del tiempo en los pacientes con influenza A (H1N1) pandémica. Varios mecanismos, incluido el barotrauma asociado con la ventilación mecánica, la toxicidad por oxígeno y la hiperinflamación, son cruciales para determinar las secuelas en estos pacientes tras recuperarse de una enfermedad grave82. Estos factores causan lesiones epiteliales leves que no se reparan adecuadamente, lo que conduce a una hiperactivación de fibroblastos, un depósito excesivo de ECM y una remodelación del parénquima pulmonar83.
No se comprende hasta qué punto la lesión mecánica y la hiperinflamación contribuyen a la fibrosis pulmonar en la COVID-19. Un número importante de pacientes graves con COVID-19 y síndrome de insuficiencia respiratoria aguda requieren cuidados críticos y asistencia respiratoria, y los que sobreviven muestran opacidades en vidrio esmerilado persistentes, disfunción pulmonar crónica y fibrosis pulmonar que afectan su calidad de vida84–86. Por otro lado, la hiperinflamación en la COVID-19 grave también incluye la liberación de quimiocinas inflamatorias, como CCL2, CCL3 e IP-10. Estos factores quimiotácticos están asociados con una activación desregulada de células del compartimento de fagocitos mononucleares, lo que podría promover aún más la hiperinflamación en pacientes con COVID-19. De hecho, el líquido bronquioalveolar de los pacientes con COVID-19 grave contiene altas concentraciones de CCL2, CCL3, CCL4 y CCL7, y una proporción disminuida de macrófagos alveolares residentes en tejidos, pero grandes cantidades de macrófagos inflamatorios derivados de monocitos87. Curiosamente, esta subpoblación de macrófagos expresa transcritos de RNA que se han asociado previamente a la reparación tisular y la promoción de la fibrosis en la cirrosis hepática88,89.
Sorprendentemente, durante la fibrosis, muchos tipos de colágeno pueden modular las funciones celulares y los procesos fisiológicos de los leucocitos y las células parenquimatosas. Además, en respuesta a la inflamación, la degradación de la ECM por las MMP genera pequeños péptidos que pueden actuar como factores quimiotácticos para los leucocitos, aumentando la inmunopatología de la enfermedad. Todos estos mecanismos promueven aún más la hiperactividad de las MMP, lo que provoca una destrucción progresiva del parénquima pulmonar90. Por lo tanto, las MMP y otros componentes de la ECM podrían actuar como lecturas de la actividad profibrótica en curso y la lesión pulmonar en pacientes con COVID-19 grave, como se analiza a continuación.
Una controversia importante que ha surgido recientemente es si la fibrosis pulmonar es una secuela exclusiva de la infección grave por SARS-CoV-2 o si los pacientes con enfermedad leve a moderada también están en riesgo. En este sentido, la evidencia de pacientes convalecientes de COVID-19, tanto adultos como pediátricos, sugiere que los pacientes mayores y aquellos con una enfermedad más grave desarrollan fibrosis91–93. De manera similar, un estudio realizado en Italia entre marzo y abril de 2020 encontró que los pacientes con manifestaciones no graves que mostraban opacidades pulmonares mostraron una remisión completa de dichas lesiones y ninguna fibrosis durante el seguimiento. Por el contrario, Dadhwal et al.94 comunicaron cinco casos de pacientes con COVID-19 asintomáticos o sintomáticos leves que presentaban disnea e imágenes del tórax que sugerían la resolución de las opacidades en vidrio esmerilado y que posteriormente desarrollaron fibrosis, 4 a 8 semanas después del diagnóstico.
A pesar de todos los estudios realizados para entender las bases inmunopatológicas de las secuelas pulmonares y extrapulmonares crónicas observadas en los pacientes con PASC o COVID-19 prolongada, su etiología aún no se comprende por completo. Una de las características esenciales de las PASC es que existe persistencia viral en diversos tejidos, como el pulmonar, el sistema nervioso central, los riñones y el intestino95,96. Se ha sugerido que los pacientes que desarrollan PASC podrían tener respuestas inmunitarias aberrantes, poco reguladas, y sobreactivación de poblaciones celulares como las de células mieloides y linfocitos T y B que infiltran en los tejidos, generando escenarios tisulares ricos en citocinas proinflamatorias o profibróticas, como es el caso de la fibrosis pulmonar post-COVID-19, caracterizada por una elevación del TGF-β y una hiperactivación de miofibroblastos productores de matriz extracelular97.
En el síndrome post-COVID-19, la disnea persistente, con frecuencia acompañada de fatiga, molestias torácicas y tos, afecta aproximadamente al 20% de los pacientes 3 meses después de la infección inicial por SARS-CoV-298. Además, un número considerable de pacientes, en particular aquellos que superan el síndrome de dificultad respiratoria aguda por COVID-19 y que requieren tratamiento con oxígeno nasal de alto flujo o ventilación mecánica, presentan secuelas pulmonares crónicas, como se demuestra mediante pruebas de función pulmonar y cambios radiológicos en la tomografía computarizada de tórax, como vidrio despulido y fibrosis99. De manera interesante, algunos estudios han mostrado que el uso de fármacos antifibróticos, como pirfenidona y nintenadib, tiene efectos clínicos y funcionales positivos para tratar secuelas pulmonares de COVID-19100,101.
Las secuelas pulmonares posvirales no son exclusivas del SARS-CoV-2 y se han descrito tras varias otras infecciones virales respiratorias, posiblemente también asociadas con alteraciones inflamatorias que conllevan daño pulmonar crónico.
En conjunto, estos datos resaltan la necesidad de realizar más estudios de investigación en pacientes convalecientes de COVID-19 para establecer mejores estrategias preventivas, terapéuticas y de rehabilitación contra la fibrosis pulmonar. Para estos fines, también se requerirán nuevos biomarcadores con valor predictivo que permitan la detección temprana de lesiones pulmonares y fibrosis.
Los mecanismos implicados en la fisiopatología de las formas graves de COVID-19 y sus secuelas se resumen en la figura 2.
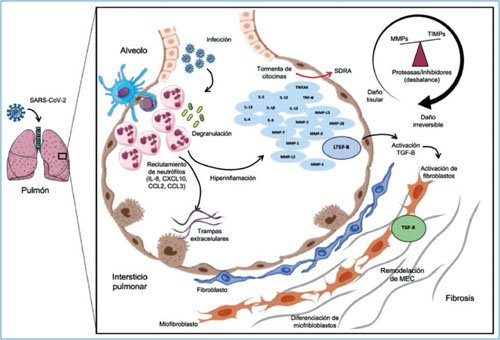
Figura 2. Inmunopatología de las formas graves de COVID-19 y sus secuelas. MEC: matriz extracelular; MMPs, metaloproteinasas de la matriz; SDRA, síndrome de dificultad respiratoria aguda; TIPMs, inhibidores tisulares de MMPs.
Conclusiones
La propagación del SARS-CoV-2 en diversos países ha provocado la posiblemente mayor crisis sanitaria global de los últimos 100 años. Aunque la gran mayoría de los casos de COVID-19 se presentan de forma asintomática o con manifestaciones leves, en ocasiones la enfermedad tiene manifestaciones graves, como neumonía viral que puede evolucionar a falla respiratoria y muerte en un periodo breve de tiempo. La evolución natural de la infección por este virus incluye una etapa inicial de infección del epitelio respiratorio y replicación viral que puede ser seguida por una segunda etapa de inmunopatología impulsada por una respuesta hiperinflamatoria que tiene manifestaciones sistémicas. El síndrome comparte características superpuestas con una linfohistiocitosis hemofagocítica inducida por virus, incluida la evidencia de activación de macrófagos con una tormenta de citocinas, y deterioro de la función de los linfocitos T y las células NK. La comprensión de la inmunopatología pulmonar y extrapulmonar de la COVID-19 permitirá identificar biomarcadores en un intento de clasificar la enfermedad como leve, moderada, grave y crítica, así como para el desarrollo de nuevas estrategias terapéuticas destinadas a reducir la hiperinflamación generalizada y pulmonar en la COVID-19 grave.
Financiamiento
El laboratorio de Inmunobiología y Genética del Instituto Nacional de Enfermedades Respiratorias Ismael Cosío Villegas recibió financiamiento del fondo E022 para investigación de la Secretaría de Salud.
Conflicto de intereses
Los autores declaran no tener conflicto de intereses.
Consideraciones éticas
Protección de personas y animales. Los autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad, consentimiento informado y aprobación ética. El estudio no involucra datos personales de pacientes ni requiere aprobación ética. No se aplican las guías SAGER.
Declaración sobre el uso de inteligencia artificial. Los autores declaran que no utilizaron ningún tipo de inteligencia artificial generativa para la redacción de este manuscrito.
